
 Обратная связь
Обратная связь4.1 The 6-month bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) regimen
This section refers to the 6-month (or 26-week) treatment regimen for MDR/RR-TB; that is, the bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) regimen. This regimen should be the initial choice for all eligible patients diagnosed with MDR/RR-TB. The recommendation in the updated WHO guidelines states:

Remarks
- Drug susceptibility testing (DST) for fluoroquinolones is strongly encouraged in people with MDR/RR-TB, and although it should not delay initiation of the BPaLM, results of the test should guide the decision on whether moxifloxacin can be retained or should be dropped from the regimen – in cases of documented resistance to fluoroquinolones, BPaL without moxifloxacin would be initiated or continued.
- This recommendation applies to the following:
- People with MDR/RR-TB or with MDR/RR-TB and resistance to fluoroquinolones (pre-XDR-TB).
- People with confirmed pulmonary TB and all forms of extrapulmonary TB except for TB involving the CNS, osteoarticular or disseminated forms of TB with multiorgan involvement.
- Adults and adolescents aged 14 years and older.
- All people regardless of HIV status.
- Patients with less than 1-month previous exposure to bedaquiline, linezolid, pretomanid or delamanid. When exposure is greater than 1 month, these patients may still receive these regimens if resistance to the specific medicines with such exposure has been ruled out.
- This recommendation does not apply to pregnant and breastfeeding women owing to limited evidence on the safety of pretomanid.
- The recommended dose of linezolid is 600 mg once daily, both for the BPaLM and the BPaL regimen.¹⁴
The BPaL regimen (bedaquiline, pretomanid, and linezolid) was first evaluated in the Nix-TB study (2015–2017, South Africa), which assessed its safety, efficacy, and pharmacokinetics for treating MDR/RR-TB, pre-XDR-TB, and patients who were intolerant or non-responsive to prior treatments. Based on promising results, the WHO recommended BPaL in 2019 under operational research conditions for MDR-TB patients with fluoroquinolone resistance and limited prior exposure to bedaquiline and linezolid.
Subsequent trials, including TB-PRACTECAL and ZeNix, further investigated BPaL-based regimens, leading to the WHO’s 2022 treatment guideline update. The TB-PRACTECAL trial was a Phase 2–3, open-label randomized controlled trial involving 419 patients across Belarus, South Africa, and Uzbekistan. Conducted by Médecins Sans Frontières (MSF) and the London School of Hygiene & Tropical Medicine (LSHTM), the trial compared three BPaL-based regimens (including BPaLM and BPaLC) against the locally approved standard of care (SoC), which was based on WHO recommendations at the time. Meanwhile, the ZeNix trial focused on optimizing linezolid dosing to improve safety and efficacy.
The work leading to the WHO’s 2022 guideline update analyzed data from these trials on several BPaL-based regimens and compared them to standard-of-care treatments and large MDR/RR-TB cohorts from South Africa and other countries. The BPaLM regimen demonstrated the highest treatment success rate (89%) in the TB-PRACTECAL trial, significantly outperforming standard regimens, which had success rates ranging from 52% to 75%. These findings solidified BPaL-based regimens as a transformative option for treating drug-resistant TB (19).
4.1.1 Eligibility
The BPaLM/BPaL regimen may be offered to patients with MDR/RR-TB in the following situations:
- pulmonary TB or most forms of extrapulmonary TB, except TB involving the CNS, osteoarticular TB or disseminated forms of TB with multiorgan involvement;
- age 14 years or older;
- no known allergy to any of the BPaLM component drugs;
- no evidence of resistance to bedaquiline, linezolid, delamanid or pretomanid, or patient has not been previously exposed to any of the component drugs for 4 weeks or longer; when exposure to the component drugs is greater than 4 weeks in duration, the patient may receive the BPaLM regimens if resistance to the specific medicines with such exposure has definitively been ruled out;
- all people regardless of HIV status;
- not XDR-TB according to the 2021 WHO definitions (20);
- not pregnant or breastfeeding.
DST for FQ is strongly encouraged in people with MDR/RR-TB; also, although it should not delay initiation of the BPaLM regimen, results of the test should guide the decision on whether moxifloxacin can be retained or should be dropped from the regimen (in cases of documented resistance to FQ, BPaL without moxifloxacin would be initiated or continued). In cases of possible FQ resistance (e.g. a history of >4 weeks of FQ use or close contact with a person infected with an FQ-resistant strain), it is best to initiate the BPaLM regimen until DST for FQ is available, to decide whether moxifloxacin should be continued, with moxifloxacin subsequently being dropped from the regimen if FQ resistance is confirmed. If the result of FQ DST is never determined or is not done, the BPaLM regimen should be used throughout. In cases of documented resistance to FQ, BPaL without moxifloxacin should be initiated or continued. If FQ resistance develops while an individual is receiving the BPaLM regimen but susceptibility to other drugs in this regimen is confirmed, moxifloxacin can be discontinued. In this scenario, BPaL can be continued because there is no benefit in retaining an ineffective drug that may cause toxicities.
Several groups of patients, as described below, were excluded from the ZeNix and TB-PRACTECAL trials. Because no data about the safety of BPaLM/BPaL in those groups are available, the inclusion of such patients should be considered with caution. The exclusions are relative, and interventions may be taken to address the underlying conditions. Once those conditions have improved or resolved, such patients can be started on BPaLM/BPaL. In some cases, the underlying condition can be managed effectively. For instance, individuals with iron deficiency anaemia and haemoglobin (Hb) levels of less than 8 g/dL can receive iron supplementation. Once Hb rises above 8 g/dL, the BPaLM/BPaL can be initiated with close monitoring and weekly complete blood count. Similarly, QT prolongation of 500 ms due to hypokalaemia can be improved by correcting potassium levels, helping to normalize the QTcF.
The following conditions should be assessed, and appropriate precautions taken:
- Patients with a very low BMI (<17 kg/m²) should be monitored closely. Although the ZeNix-TB trial excluded those with a BMI of less than 17 kg/m², TB-PRACTECAL had no such exclusion criterion and 167 (40%) of those in TB-PRACTECAL had a BMI of less than 17 kg/m². Low BMI should not be an absolute contraindication when commencing the BPaLM/BPaL regimen. In the multicountry operational research study, a cohort of 71 individuals with a BMI lower than 17 kg/m² enrolled in the BPaL regimen achieved a success rate of 94% (21).
- Linezolid is associated with peripheral neuropathy; therefore, those with preexisting peripheral neuropathy of Grade 3–4 should be considered carefully before commencing the BPaLM/BPaL regimen. These advanced grades of neuropathy cause significant disability, which may be exacerbated by the use of linezolid. Alternatively, to decrease the risk of peripheral neuropathy exacerbation, a 9-month linezolid-sparing regimen could be used.
- Linezolid is also associated with anaemia, neutropenia and thrombocytopenia, and caution is advised in patients with these conditions, particularly in those with a Hb level of less than 8 g/dL, a neutrophil count of less than 750/mm3 or a platelet count of less than 150 000/mm3 (See Annex 2).
- Patients with liver enzymes at levels three times greater than the upper limit of normal were excluded from both the ZeNix and TB-PRACTECAL trials, because bedaquiline and pretomanid are both associated with increases in liver enzymes. Other causes of transaminase elevation should be ruled out (e.g. concomitant hepatotoxic medicines for other comorbidities or CLD). In such patients, BPaLM/BPaL may be started when the liver enzymes improve.
- Known history of cardiac disease – including baseline corrected QT interval by Fridericia (QTcF) of more than 500 ms – raises concern, particularly in individuals with syncopal episodes, significant arrythmias, personal or family history of congenital QTc prolongation, torsade de pointes (tdP), bradyarrhythmia or cardiomyopathy. Both bedaquiline and moxifloxacin can prolong QT interval. While reports of serious AEs and mortality associated with QTc prolongation are rare, caution is strongly advised.
Where patients are moribund or have advanced TB disease, symptom control and palliative care may be more appropriate than initiation of treatment. Decisions should be guided by clinical judgement and the patient’s preferences.
4.1.2 Composition, dosing and duration of the regimen
Composition
The BPaLM regimen comprises four medications: bedaquiline, pretomanid, linezolid and moxifloxacin. In contrast, the BPaL regimen includes three of those medications but excludes moxifloxacin, making it suitable for patients with confirmed FQ resistance.
Pretomanid, the new drug in the regimen, is a nitroimidazole and a prodrug that is metabolically activated by a nitroreductase, producing various metabolites that are responsible for its therapeutic action. Pretomanid acts as a bactericidal agent by inhibiting cell wall biosynthesis; thus, it has an excellent sterilizing capacity. Further information about the mechanism of action and AEs of each drug can be found in Annex 1.
Dosing
The same doses for bedaquiline, pretomanid and linezolid are used in the BPaLM/BPaL regimen. Bedaquiline is dosed at 400 mg once daily for 2 weeks, then 200 mg three times per week afterwards, according to the product label. However, in the ZeNix trial, bedaquiline was administered at 200 mg daily for 8 weeks followed by 100 mg daily. Pharmacokinetic simulations showed that administration of bedaquiline as daily dosages was comparable to the labelled regimen (400 mg daily for 14 days followed by 200 mg three times a week) (22). Any difference was marginal and unlikely to be expressed as a meaningful difference in response. This daily bedaquiline dosing regimen was evaluated in the ZeNix trial (23) and is being further evaluated in the SimpliciTB trial (24, 25). Such dosing may be more convenient for patients and health care providers because it allows for daily dosing of all drugs throughout the regimen and thus may improve adherence. Pretomanid is administered at 200 mg once daily (see Table 2.4.1). Linezolid dosing is administered at 600 mg once daily and moxifloxacin at 400 mg once daily (in the BPaLM regimen).
Table 2.4.1. Dosing of component drugs for adults and adolescents (aged ≥14 years) for BPaLM and BPaL
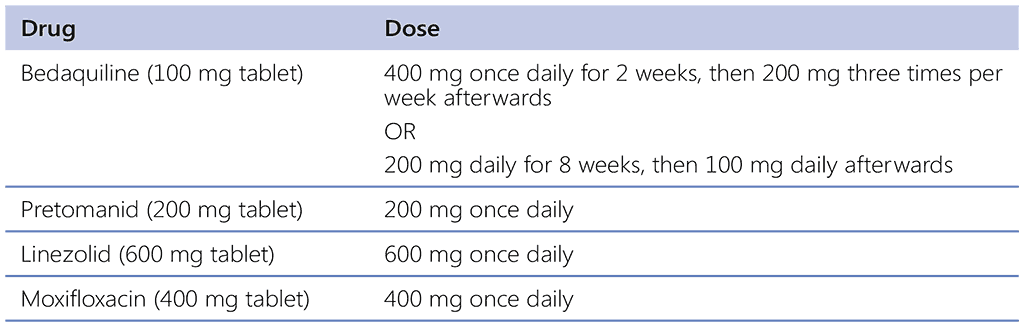
BPaLM: bedaquiline, pretomanid, linezolid and moxifloxacin.
It is preferable to continue linezolid at the full dose for the entire duration of the regimen. However, the dose of linezolid can be reduced to 300 mg or the drug can be discontinued if there is significant toxicity (depending on the severity of specific AEs or serious AEs) associated with linezolid, including optic neuritis, peripheral neuropathy or myelosuppression; the drug can be restarted later where possible. Dose modification of linezolid should be avoided during the first 9 weeks of therapy if possible. Further considerations on linezolid dosing are discussed below in Section 4.2.4. Dose modifications for bedaquiline, moxifloxacin and pretomanid are not recommended. Given the lack of evidence for the use of other FQ, the GDG was unable to recommend the substitution of moxifloxacin with levofloxacin.
In the ZeNix trial, all medications were administered with food throughout, because the bioavailability of bedaquiline (and pretomanid) increases when taken with meals. Therefore, it is preferable that the BPaLM/BPaL regimen is administered with food.
Duration
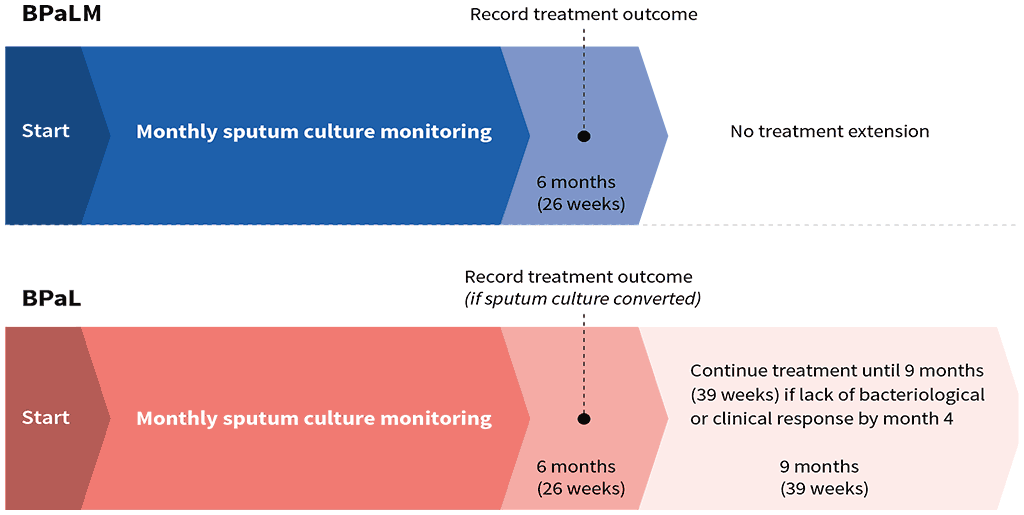
During the GDG meeting, the slight differences in the treatment duration of the BPaLM and BPaL regimens, as studied in the TB-PRACTECAL and ZeNix trials, were acknowledged and discussed, and the panel suggested standardizing treatment duration of BPaLM to 6 months (26 weeks) during programmatic implementation. For BPaL, the GDG suggested the possibility of an extension to a total of 9 months (39 weeks) if no culture conversion or clinical response by month 4 (Fig. 2.4.1). All medicines in the regimen would be used throughout the treatment, including the extension from 26 to 39 weeks (when BPaL is used). Ideally, missing doses of all three or four drugs in the regimen should be avoided; however, if doses are missed, any interruption of longer than 7 days should be made up for by extending the treatment duration (for the number of missed doses); therefore, 26 or 39 weeks of prescribed doses should be completed within an overall period of 7 or 10 months, respectively.
Patients with susceptibility to FQ can be started on the BPaLM regimen for 6 months (26 weeks). In the case of resistance to FQ at the baseline that is identified after treatment initiation, moxifloxacin may be discontinued and the regimen can be continued as BPaL. When the regimen is BPaL from the start or is changed to BPaL, it can also be extended to a total of 9 months (39 weeks), continuing from the start of the therapy with a BPaL-based regimen. This extension of the BPaL regimen would be needed only in cases where there is a lack of culture conversion or clinical response (based on the radiological response and clinical judgement of the treating physician) by month 4. Close monitoring of treatment response is necessary for patients needing treatment extension. Once a decision has been made to extend treatment beyond 6 months, further sputum culture results must be followed up, to ensure that culture conversion occurs soon after month 4 on treatment. If the month 5 and 6 culture results remain persistently positive, treatment failure should be suspected, particularly if the patient has had suboptimal adherence to treatment or shows other signs of poor clinical or radiological response to treatment. This includes conducting DST to detect any acquired drug resistance to the component medicines. Extension of BPaL to 9 months should be undertaken with caution in patients with a high number of missed linezolid dosages – switching to an individualized longer regimen may be considered instead of a BPaL extension.
For individuals who switch from BPaLM to BPaL, the treatment start date should be considered as the date BPaLM was initiated, because the patient remained on treatment with three effective drugs during the entire treatment period.
Fig. 2.4.1 Duration of treatment with BPaLM and BPaL regimen options
4.1.3 Modifications of treatment
Linezolid modification
Linezolid is by far the most toxic drug in the BPaL-based regimens and is among the TB medicines with the highest incidence of adverse events (26). Although it is preferred to continue linezolid at the full dose for the entire duration, the dose may need to be reduced to 300 mg or discontinued (and restarted when possible) if there is significant toxicity.
Linezolid is associated with myelosuppression (anaemia, neutropenia and thrombocytopenia), with peripheral neuropathy and optic neuropathy, and the AEs were noted more frequently in regimens using high-dose linezolid (1200 mg daily). In the ZeNix study, 31% (14/45) of participants in the study arm using linezolid 1200 mg daily for up to 26 weeks experienced an AE of Grade 3 or more compared with 20% (9/45) of participants in the intervention arm using 600 mg daily of linezolid for 26 weeks, and 19.6% (20/102) of participants in the BPaL arm using linezolid 600 mg or 300 mg daily in the TB-PRACTECAL trial. In the multicountry BPaL operational research study, AEs of Grade 3 or higher were noted in 25.3% (75/297) of participants among those started with linezolid 1200 mg per day and 9.4% (5/53) among those started with linezolid 600 mg per day (21).
The most common AEs resulting in dose modifications or early interruption of linezolid in the Nix-TB study (1200 mg dose of linezolid used) were peripheral neuropathy (81%) and myelosuppression (48%), but these AEs were often not severe and they improved with dose reductions or cessation of the drug. A toxicodynamic modelling study using Nix-TB data showed lower percentages of patients with severe peripheral neuropathy (median 5% versus 19%) and severe anaemia (1% versus 15%) in patients using 600 mg daily compared with 1200 mg linezolid daily (27). Hence, care should be taken for patients who have an Hb level of less than 8 g/dL or a platelet count of less than 150 000/mm³.
Myelosuppression (even of Grade 3 or 4) is often reversible with a short (e.g. 1-to-2-week) break from linezolid followed by resumption of the drug at a reduced dose of 300 mg per day, if needed, or to 600 mg per day, as tolerated; severe anaemia may need to be treated with blood transfusion.
Although optic neuritis was infrequent, it is an important AE that can result in the permanent discontinuation of linezolid. In the Nix-TB study, two patients had optic neuritis during the fourth or fifth month of the BPaL regimen using 1200 mg per day, and symptoms resolved in both patients after permanent discontinuation of the regimen. In the LIFT-TB study, optic neuritis occurred in two patients given 600 mg of linezolid per day, and both improved and resolved after discontinuation.
In the ZeNix and TB-PRACTECAL trials, more than 90% of participants were able to complete more than 75% of the maximal intended dose and duration of linezolid. In the multicountry operational research study, in the 600 mg daily dose group (n=53), 25% had linezolid dose modification due to AE (6 patients with linezolid dose reduction, 2 with interruption and 5 with permanent discontinuation). Despite these modifications, treatment success was 84.6% (1 person died and 1 was not evaluated) (21).
Actions should be taken either before starting treatment or during treatment with BPaL-based regimens to address the common toxicities associated with linezolid. Such actions include dose reduction to 300 mg per day, temporary interruption after 9 weeks of 600 mg per day or permanent discontinuation after 18 weeks of treatment.
Examples of changes in linezolid dosing are given in Box 2.4.1.

Modifications of linezolid dose were made in the Nix-TB, ZeNix and TB-PRACTECAL trials when toxicity associated with linezolid was suspected. Although no analysis was undertaken to determine whether individuals with dose reductions had poorer treatment outcomes than those who continued 600 mg daily for the complete duration, overall treatment success was high in all investigational arms.
Dose modification and temporary interruption of linezolid is acceptable after the first 9 weeks of treatment with 600 mg of linezolid per day in case of AEs. However, this principle should not override the need to avoid permanent disabilities. In some circumstances, linezolid may need to be permanently stopped and a decision made on whether to continue the other drugs to complete treatment or start a new treatment. After 9 weeks of consecutive administration of 600 mg of linezolid per day, the dose of linezolid can be reduced to 300 mg, if necessary (see examples in Box 2.4.2) or the drug may be ceased for 1–2 weeks.
Pharmacokinetic studies have suggested that dose optimization of linezolid may differ among patients (28). Therapeutic drug monitoring (TDM) is a novel approach that can be used, where available, to optimize linezolid dose and minimize AEs, without compromising effectiveness (29).
Further research is encouraged and is ongoing, to help ascertain how to optimize TDM in the treatment of individuals with MDR/RR-TB who are prescribed linezolid.
The findings from a dosing strategy study evaluating Nix-TB study data suggested that monitoring neuropathy symptoms and Hb levels may help to guide linezolid dosing to avoid toxicities. A decrease in Hb level of 10% or more after 4 weeks of treatment may help to identify those at high risk for severe anaemia (27). However, further research on dosing strategies is needed to optimize when and how to reduce the dose of linezolid.
Modification or discontinuation of full BPaLM/BPaL regimen
The only experience using the full BPaLM/BPaL regimens stems from two clinical trials; hence, it is suggested that the programmatic implementation be aligned with this experience. Safe management of AEs may warrant dose reduction or discontinuation of the component drugs. However, the BPaLM/BPaL regimen has been studied as a standardized course of treatment with allowable modifications at certain points in the treatment course (Table 2.4.2). Modification of the regimen outside the recommended durations or replacement of any of the component drugs may result in poor treatment outcomes.
Temporary cessation of full BPaLM/BPaL regimen
Dose modification of bedaquiline and pretomanid is not recommended; rather, temporary cessation of the full regimen is allowed for suspected drug-related toxicity for the following durations:
- not more than 2 weeks of consecutive treatment interruption of all medicines in the regimen; or
- not more than 4 weeks cumulative of non-consecutive treatment interruption of all medicines in the regimen.
Reintroduction of the full regimen could be considered after the temporary cessation. Missed doses need to be made up and added to at the end of treatment.
The durations given above for temporary cessation of the full regimen provide general guidance for conducting a clinical review and possibly changing to an individualized longer regimen.
Permanent discontinuation of BPaLM/BPaL and change to another regimen
The BPaL-based regimen may need to be permanently discontinued in some patients for whom the interruption goes beyond the recommended duration. In such cases, patients need to be evaluated and their treatment switched to an individualized longer regimen, based on the WHO guidelines for regimen design using priority grouping of medicines. The most common situations in which the regimen may be discontinued and shifted to another regimen are treatment failure, inability to use linezolid for enough time owing to AEs (see Table 2.4.2), or pregnancy that occurs during treatment.
As an exception, patients who have started a different regimen (e.g. BPaL or BPaLM) may change to one of the BDLLfxC regimens if they can no longer use or access pretomanid (e.g. if a patient becomes pregnant after starting BPaL or BPaLM, or if pretomanid drug supply is interrupted). In such scenarios, the bedaquiline dosing schedule from the BPaLM regimen may be carried over into the BDLLfxC regimen to prevent excessively prolonged treatment durations that could arise from adding the full duration of the new regimen. While this approach may be logical and patient-centered, it is important to note that there is no available evidence to support it. Each case must be carefully reviewed, and clinical judgment should be applied on an individual basis.
Table 2.4.2. Modifications of treatment with the BPaLM/BPaL regimen
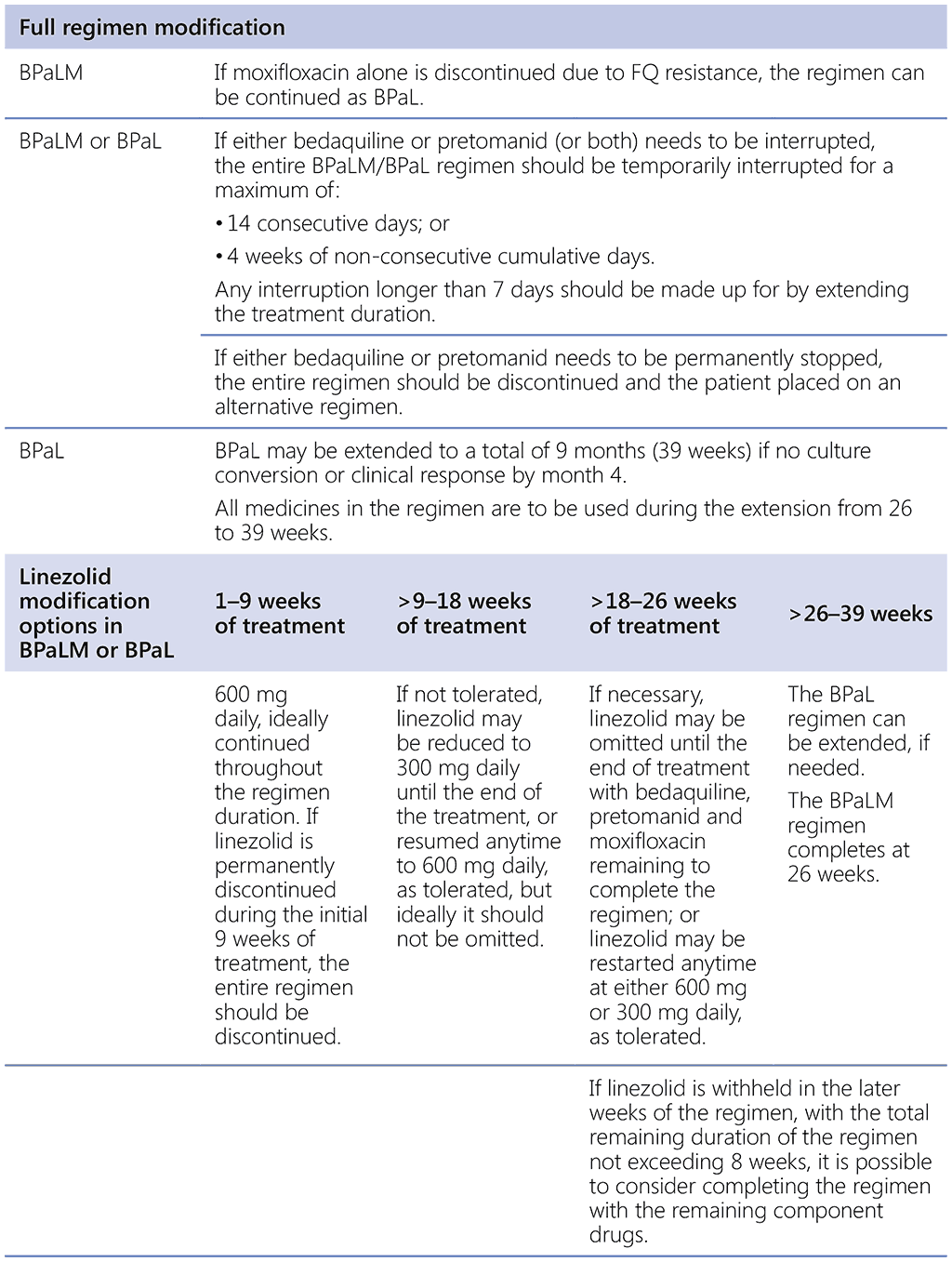
BPaL: bedaquiline, pretomanid and linezolid; BPaLM: bedaquiline, pretomanid, linezolid and moxifloxacin; FQ: fluoroquinolones.

4.1.4 Key subgroups
The following subgroup analyses were available to the GDG in considering the eligibility criteria for the BPaLM/BPaL regimen: age, smear status, smoking status, pulmonary TB patients with radiological evidence of bilateral disease or radiological evidence of cavitation, PLHIV and previous TB. The GDG also considered the eligibility criteria as stipulated by each of the trials. Several groups of patients were excluded from the ZeNix and TB-PRACTECAL trials (e.g. extrapulmonary TB patients, pregnant or breastfeeding women, children and adolescents < 14 years). Inclusion of such patients should be considered with caution, because there are currently no available data regarding the safety of BPaLM/BPaL in such populations. This subsection summarizes the resulting considerations when prescribing the BPaLM/BPaL regimen.
Extensive pulmonary TB
The Nix-TB, ZeNix and TB-PRACTECAL studies included pulmonary TB patients with radiological evidence of bilateral disease or radiological evidence of cavitation. Hence, patients with extensive pulmonary disease can be started on the BPaLM/BPaL regimen; however, close microbiological and clinical monitoring for culture conversion and clinical or radiological response should be maintained.
Extrapulmonary TB
WHO recommends the BPaLM/BPaL regimen for extrapulmonary TB, except for TB involving the CNS, osteoarticular TB and disseminated forms of TB with multiorgan involvement. The longer MDR-TB regimens apply to such patients. There is a single case study of a participant from the Nix-TB study that confirms the incidental use of BPaL in CNS TB with a favourable outcome (30), but further studies are required to recommend the regimen for programmatic implementation in severe disseminated TB. Linezolid has excellent penetration of the cerebrospinal fluid (CSF) and brain, and studies involving a small number of participants indicated that bedaquiline penetrates well into the CSF, but there are no data on pretomanid on CSF or brain penetration (Annex 1).
People living with HIV
PLHIV represented 19.5% of those enrolled in the ZeNix trial, 51% of those enrolled in the Nix-TB study, and 26.7% of those enrolled in the TB-PRACTECAL trial. These patients were eligible to enrol in the ZeNix trial if they had a CD4 count of more than 100 cells/mm³, and in TB-PRACTECAL regardless of CD4 count. Thus, patients can be enrolled in the BPaLM/BPaL regimen irrespective of the CD4 count; however, care should be taken when CD4 counts are below 100 cells/mm3. In the LIFT-TB study, 14 patients had PLHIV, of whom 13 received linezolid 1200 mg daily. Treatment success was 92.9% (13/14); only one patient who was given 1200 mg per day failed treatment.
In the ZeNix trial, enrolled participants had to be taking permitted ARV medications.¹⁵ Although there was no such criterion in TB-PRACTECAL, it is important to consider DDIs when administering TB and HIV medications in combination (see Section 4.1.1). The ARV drug efavirenz induces the metabolism of bedaquiline, so its co-administration with bedaquiline may result in reduced bedaquiline exposure and loss of activity; therefore, co-administration is to be avoided. Efavirenz also reduces pretomanid exposures significantly (31); therefore, an alternative ARV agent (potentially dolutegravir, although there is currently insufficient evidence for this) should be used if the BPaLM/BPaL regimen is considered. Ritonavir increases bedaquiline exposure, which could potentially increase the risk of bedaquiline-related adverse reactions (32); however, increased risk has not been noted in studies administering both drugs concurrently (32–34), including in the ZeNix trial. Individuals who are prescribed both bedaquiline and ritonavir should be monitored closely for AEs, including QTc prolongation. Finally, ARV regimens including zidovudine should be avoided, if possible, because both zidovudine and linezolid may cause myelosuppression (more details in Annex 2).
Pregnant and breastfeeding women
Pregnant and breastfeeding women were excluded from both the ZeNix and TB-PRACTECAL trials; hence, no analysis specific to this subgroup of patients could be performed, and the safety of the BPaLM/BPaL regimen (especially of pretomanid) in pregnant and breastfeeding women has not been established. For pregnant and breastfeeding women, the 6-month regimen BDLLfxC or 9-month regimens should be considered (Sections 4.2 and 5).
Children and adolescents
Children and young adolescents were excluded from the Nix-TB, ZeNix (0–13 years) and TB-PRACTECAL (0–14 years) studies; therefore, it was not possible to perform an analysis specific to this subgroup of patients. Although bedaquiline, linezolid and FQ have been used to treat MDR/RR-TB in younger patients, there are no data about the use of pretomanid in this population, and further study is required to expand the use of BPaLM/BPaL to these patients.
Individuals aged 14 years and older were included in the Nix-TB and ZeNix trials, and individuals aged 15 years and older in TB-PRACTECAL; thus, such individuals can safely be started on the BPaLM/BPaL regimen under programme conditions. It is recommended that younger patients aged below 14 years with pulmonary MDR/RR-TB be given consideration for the newly recommended 6-month BDLLfxC regimen or 9-month regimens.
4.1.5 Implementation considerations
DST and resistance to the component medicines
DST for FQ is strongly encouraged in people with MDR/RR-TB. A WHO-recommended rapid molecular test should be used as an initial rapid test (in preference to culture and phenotypic DST) to detect resistance to FQ (35). The results of the DST, when available, should guide the decision on the use or changes of this regimen; however, initiation of the BPaLM regimen should not be delayed by the wait for these results.
For patients who submit a sputum sample for culture-based second-line DST at the beginning of treatment, results may not be available until after treatment has started. If resistance to any of the regimen component drugs (except moxifloxacin) is discovered after treatment has been initiated, the regimen needs to be discontinued. Although DST for bedaquiline, pretomanid and linezolid is not easily accessible, in situations where it is available, it is highly desirable to carry this out at baseline. Patients with strains resistant to bedaquiline, pretomanid or linezolid should commence treatment with either a 9-month regimen or a longer, individualized MDR-TB regimen.
It is critical to know the resistance profile of the patient and thus exclude resistance to the anti-TB drugs composing the BPaLM/BPaL regimen. Molecular (genotypic) methods have considerable advantages, in particular with regard to their speed, the standardization of testing, their potentially high throughput and the reduced requirements for biosafety.
Many different rapid molecular DST methods are available for moxifloxacin (LPA, LC-aNAATs and targeted NGS). If resistance to moxifloxacin is detected, then moxifloxacin should be discontinued and the patient should be continued with the BPaL regimen.
Only one molecular method, targeted NGS, is available to detect resistance to bedaquiline and linezolid; where this method is available, it should be performed before the start of the BPaLM/BPaL regimen. The results of targeted NGS for bedaquiline and linezolid should be critically assessed, taking into account the prevalence of resistance to these drugs in tested populations and the risk of resistance in a particular patient.
Certain individuals are at high risk for resistance to bedaquiline and linezolid; for example, people with prior drug exposure, populations where the prevalence of resistance is above 5% and those with a history of contact with a person with TB with known drug resistance. For such individuals, a negative (sensitive) result of the targeted NGS phenotypic testing is advised. In addition, if resistance to bedaquiline is detected (in cases of low or high risk of this resistance in particular patients), the use of the phenotypic DST is still advised.
Finally, the critical concentrations for pretomanid phenotypic resistance detection have been established for the MGIT method, enabling NTPs to perform phenotypic DST.
If resistance to bedaquiline, pretomanid or linezolid is detected in patients with MDR/RR-TB, the BPaLM/BPaL regimen should not be offered, and the patient must switch to another appropriate treatment regimen.
If resistance to bedaquiline, linezolid or pretomanid develops while on the regimen, the treatment is considered to have failed, and individuals should be shifted to either a 9-month regimen or a longer individualized regimen (Sections 5 and 6).
In the absence of DST for bedaquiline, pretomanid and linezolid, treatment decisions will rely on the likelihood of effectiveness of these medicines, based on an individual patient’s clinical history of exposure to the drugs and DRS data from the country or region. This should be considered an interim measure until the DST capacity for these drugs becomes available.
Further details on treatment modifications and follow-on DST for MDR/RR-TB patients based on results from targeted NGS can be found in the operational handbook Module 3: Diagnosis – rapid diagnostics for tuberculosis detection (11).
History of exposure to component drugs
As detailed previously, DST for FQ is advisable before starting therapy. Where DST is unavailable, a careful history of previous exposure to TB therapy is critical. Where the treatment history suggests there may be resistance to one of the components of the regimen (i.e. exposure to bedaquiline, pretomanid or linezolid in an inadequate regimen for more than 1 month) and DST is unavailable, the BPaLM/BPaL therapy should not be commenced; instead, a longer individualized regimen should be considered. On the other hand, if there is greater than 1 month exposure to FQ that indicates probable resistance, the BPaLM regimen may be initiated and continued until resistance to FQ is confirmed.
Previous exposure to delamanid may suggest cross-resistance to pretomanid (36) and exposure to clofazimine may suggest cross-resistance to bedaquiline (37). Caution should be exercised in such situations and, where possible, if DST for clofazimine, bedaquiline, pretomanid or delamanid is available, it should be used. However, the BPaLM regimen can be started in these situations and the treatment will be reviewed once the DST result to these drugs is available.
Drug–drug interactions
Vigilance or, preferably, drug substitution should be considered when certain medications are prescribed concurrently with the BPaLM/BPaL regimen. Common DDIs of the regimen components with other concomitant medicines are described in detail in Annex 1 and Annex 2. In each of the above situations, if the clinician judges that the potential benefits outweigh the potential risk (also considering alternative treatment options), then treatment may proceed with caution.
Cost and cost–effectiveness analysis
A 6-month BPaLM treatment course for an MDR/RR-TB patient costs US$ 364 compared with US$ 926–998 for the 6-month BDLLfxC regimen, and US$ 209–1500 for the 9-month regimens. For a patient with pre-XDR-TB, a 6-month course of BPaL costs US$ 340 versus the cost of an 18-month treatment regimen (with no documented resistance to the Group A drugs, bedaquiline and linezolid) of US$ 453 (38).¹⁶
Although there have been no cost–effectiveness studies looking at the BPaLM regimen, such studies for the BPaL regimen have demonstrated lower costs. Owing to the substantially shorter treatment duration and reduced need for hospitalization (39, 40), it was estimated that the implementation of a national programme using BPaL for MDR/RR-TB would cost 57–78% less than conventional longer regimens, when including all costs (e.g. investigations, drugs and hospitalization) (39). With the decrease in the cost of bedaquiline and pretomanid in 2025, these analyses are becoming even more favourable for the implementation of the BPaLM/BPaL regimen.
Treatment support
Measures to support patient adherence tailored to patient needs are important to retain patients on treatment and ensure good treatment outcomes. Support should be provided through an effective model of care and measures should include support in the community or at home, social support and digital health interventions for communication with the patient (41) (see more details in Chapter 3). Early ambulatory care was employed by the ZeNix trial and is recommended for the programmatic implementation of the BPaLM/BPaL regimen, because it complements the patient-centred approach to the management of TB. Patient support is particularly important for the BPaLM/BPaL regimen, to ensure adherence and avoid treatment interruption and LTFU. Interruption of all medicines is especially dangerous for the BPaLM/BPaL regimen and all other bedaquiline-based regimens because it effectively leaves patients on bedaquiline monotherapy owing to the long half-life of bedaquiline.
4.2 The 6-month bedaquiline, delamanid, linezolid, levofloxacin and clofazimine (BDLLfxC) regimen
This section refers to the use of BDLLfxC – a new 6-month regimen containing bedaquiline, delamanid, linezolid, levofloxacin and clofazimine – for the treatment of MDR/RR-TB. This short regimen is particularly appropriate for people who are unable to benefit from the currently recommended 6-month BPaLM regimen owing, for example, to restricted access to pretomanid, being aged younger than 14 years, or being pregnant or breastfeeding. The recommendation in the updated 2025 guideline states:

Remarks
- Drug susceptibility testing (DST) for fluoroquinolones is strongly encouraged in people with MDR/RR-TB. Although it should not delay the initiation of the BDLLfxC, the test results should guide the decision on whether levofloxacin or clofazimine should be retained.
- This recommendation applies to the following:
- People with MDR/RR-TB or pre-XDR-TB (i.e. MDR/RR-TB and resistance to fluoroquinolones).
- Patients with less than 1 month of previous exposure to bedaquiline, linezolid, delamanid or clofazimine. When exposure is greater than 1 month, these patients may still receive the regimen if resistance to the specific medicines involved in such exposure has been ruled out.
- People with diagnosed pulmonary TB of all ages, including children, adolescents, PLHIV, and pregnant and breastfeeding women.
- People with all forms of extrapulmonary TB except for TB involving the CNS, osteoarticular TB, or disseminated forms of TB with multiorgan involvement.
- Children and adolescents who do not have bacteriological confirmation of TB or resistance patterns but who do have a high likelihood of MDR/RR-TB (based on clinical signs and symptoms of TB, in combination with a history of contact with a patient with confirmed MDR/RR-TB).
- When resistance to fluoroquinolones is unknown, the regimen should be started as BDLLfxC and then adjusted based on the DST results. In cases of quinolone susceptibility, clofazimine should be dropped and the regimen started or continued as bedaquiline, delamanid, linezolid and levofloxacin (BDLLfx). In cases of resistance to fluoroquinolones, levofloxacin should be dropped and the regimen started or continued as bedaquiline, delamanid, linezolid and clofazimine (BDLC).
Since the release of the previous WHO DR-TB guidelines in December 2022 (42), data on a new 6-month oral regimen for MDR/RR-TB and pre-XDR-TB have become available from the BEAT Tuberculosis clinical trial in South Africa (43). This was a pragmatic, open-label, RCT conducted at two government health facilities in different provinces between August 2019 and September 2022. The trial used a non-inferiority design to compare safety and efficacy of the study strategy with the SoC approach in South Africa (44). People presenting to the hospital sites for diagnosis and treatment of MDR/RR-TB were asked to give written informed consent for trial participation and were then randomized to the study strategy (6 months of bedaquiline, delamanid, linezolid and either levofloxacin or clofazimine, or both) or the control strategy (WHO-recommended, mostly 9-month all-oral, bedaquiline-containing regimen for FQ-susceptible TB and longer regimen for FQ-resistant TB patients). The study strategy used a “tapering down” approach, whereby participants diagnosed with TB that was resistant to at least rifampicin would initiate a regimen composed of bedaquiline, delamanid, linezolid, levofloxacin and clofazimine. Once FQ DST results became available, either immediately or later, the regimen was altered as follows: clofazimine was withdrawn (and levofloxacin continued) when FQ susceptibility was confirmed, levofloxacin was withdrawn (and clofazimine continued) when FQ resistance was confirmed, and neither drug was withdrawn (both levofloxacin and clofazimine continued) in cases where FQ DST results were unavailable. This tapering down approach made provision for the potential delay in obtaining FQ DST results while still allowing rapid initiation of effective MDR/RR-TB treatment, which provided adequate cover for both FQ-susceptible and FQ-resistant strains from the outset.
The evidence for the novel 6-month BDLLfxC regimen that was used to inform PICO questions for the updated WHO guidelines was derived from BEAT Tuberculosis RCT. However, an unrelated study with a similar name – the BEAT-India (Building Evidence to Advance Treatment of TB) study – was a prospective open-label, single-group cohort study that was carried out at five sites in India between April 2019 and January 2021 (45). The BEAT-India study only enrolled patients with pre-XDR-TB, but the 6-month study regimen (BDLC) was the same as that used for participants with pre-XDR-TB enrolled in the study strategy of the BEAT Tuberculosis trial. Although not considered in the PICO questions for these guidelines, the encouraging findings from the BEAT-India study also appear to support the recommendations for implementation of a 6-month regimen containing these medicines in settings outside of South Africa.
The BEAT Tuberculosis trial enrolled 402 participants: 202 to the study strategy and 200 to the control strategy. Most participants in the control strategy received the WHO-recommended 9-month regimen containing bedaquiline and linezolid. The study protocol allowed for enrolment of PLHIV, children aged over 6 years, adolescents, and pregnant and breastfeeding women. Patients with extensive or severe pulmonary TB were also included, as were people with non-severe extrapulmonary forms.
The results of this RCT support the programmatic use of a 6-month regimen containing bedaquiline, delamanid and linezolid, as well as levofloxacin or clofazimine (or both), in place of 9-month or longer regimens in patients with MDR/RR-TB or pre-XDR-TB who are unable to receive pretomanid within the BPaL or BPaLM regimens.
4.2.1 Eligibility
The 6-month BDLLfxC regimen may be offered in the following situations:
- people with MDR/RR-TB or pre-XDR-TB;
- people with MDR/RR-TB and less than 1 month of previous exposure to bedaquiline, linezolid, delamanid or clofazimine; when exposure is greater than 1 month, these patients may still receive the regimen if resistance to the specific medicines with such exposure has been ruled out;
- people with diagnosed pulmonary TB, including children, adolescents, PLHIV, pregnant and breastfeeding women;
- people with most forms of extrapulmonary disease except for TB involving the CNS, or osteoarticular or disseminated forms of TB with multiorgan involvement; and
- children and adolescents who do not have bacteriological confirmation of TB or resistance patterns but who do have a high likelihood of MDR/RR-TB (based on clinical signs and symptoms of TB, in combination with a history of contact with a patient with MDR/RR-TB).
Note that children, adolescents and adults with any extent or severity of pulmonary TB disease¹⁷ are eligible to receive this 6-month regimen. In addition, people with non-severe forms of extrapulmonary TB, such as uncomplicated pleural effusions or peripheral lymph node disease, are also eligible to receive this 6-month regimen.
The 6-month BDLLfxC regimen may also be offered to adults (who might otherwise have been eligible for BPaL or BPaLM) in settings where pretomanid is restricted, unavailable or contraindicated (e.g. in pregnancy or breastfeeding), or to patients who are unable to tolerate pretomanid.
The 6-month regimen is NOT appropriate for the following situations:
- patients with MDR/RR-TB with documented resistance to bedaquiline, delamanid, linezolid or clofazimine;
- people in whom the most recent episode of MDR/RR-TB treatment has failed; or
- people with severe extrapulmonary TB.¹⁸
4.2.2 Composition, dosing and duration of the regimen
The initial diagnosis of MDR/RR-TB is usually based on the results of mWRDs. DST for FQ is strongly encouraged in people with MDR/RR-TB, because the test results will guide the decision as to whether levofloxacin or clofazimine should be retained or dropped from the regimen. However, FQ DST results are not always immediately available at the time of diagnosis. For people who are eligible to receive this 6-month regimen, the absence of the FQ DST result should not delay initiation of treatment with a regimen containing all five drugs (i.e. bedaquiline, delamanid, linezolid, levofloxacin and clofazimine). The regimen may then be adjusted by withdrawal of either levofloxacin or clofazimine (or neither), depending on whether FQ DST was successful, and whether the result showed FQ resistance or susceptibility. Therefore, patients receiving this 6-month regimen are treated with either BDLLfxC, BDLLfx or BDLC.
These regimen variations should be given for a total of at least 24 weeks (i.e. 6 months). In some situations, e.g. lack of bacteriological or clinical response by month 4, treatment may be extended up to a total of 9 months (see section 4.2.5). If the month 5 and 6 culture results remain persistently positive, treatment failure should be suspected, particularly if the patient has had suboptimal adherence to treatment or shows other signs of poor clinical or radiological response to treatment.
Ideally, all component medicines of each regimen variation should be used for the full duration of treatment, but in some situations (described below), linezolid may have to be discontinued prematurely.
Composition
BDLLfxC regimen
People with MDR/RR-TB who meet the eligibility criteria may start treatment with the BDLLfxC regimen, then modify to either BDLLfx or BDLC once FQ DST results are available, as described below.
In cases where DST for FQ is unknown, or cannot be carried out at baseline, the regimen will include all five medicines – bedaquiline, delamanid, linezolid, levofloxacin and clofazimine – for the duration of treatment. If FQ DST can be repeated on a clinical sample taken at some point after starting treatment, the regimen may still be adjusted later according to the DST results, as described below.
BDLLfx regimen
In cases where the MDR/RR-TB strain is shown to be susceptible to FQ on DST, the regimen will include four medicines – bedaquiline, delamanid, linezolid and levofloxacin (i.e. BDLLfx).
If the DST result is available before the start of treatment and proves susceptibility to FQ, patients may commence the four-drug BDLLfx regimen immediately. If the DST result is delayed, the patient will initiate the BDLLfxC regimen and, once the DST result confirms FQ susceptibility, clofazimine can be ceased and BDLLfx continued for the remainder of treatment.
It is possible for a patient with confirmed FQ-susceptible MDR/RR-TB, who is receiving a BDLLfx regimen, to switch to BDLC if necessary; for example, intolerance to levofloxacin, limited availability of levofloxacin, or patient or provider preference.
BDLC regimen
In cases where the MDR/RR-TB strain is shown to be resistant to FQ on DST, the regimen will include four medicines – bedaquiline, delamanid, linezolid and clofazimine (i.e. BDLC).
If the DST result is available before the start of treatment and proves resistance to FQ, patients may start on the four-drug BDLC regimen immediately. If the DST result is delayed, the patient will initiate the BDLLfxC regimen; if the DST result then confirms FQ resistance, levofloxacin can be dropped and BDLC continued for the remainder of treatment.
There is a potentially higher concern for further drug resistance in strains of pre-XDR-TB; thus, every effort must be made in these cases to ensure that DST is undertaken as quickly as possible for other component drugs in the regimen. Detection of resistance to any of the drugs in this regimen will necessitate a rapid switch to another appropriate regimen.
Dosing
Dosing of medicines in the regimen for both adults and children should follow the latest WHO weight-based dosing tables (see Annex 4). All reasonable efforts should be made to ensure the availability of child-friendly formulations.
Bedaquiline
Bedaquiline may be dosed in one of two ways in this regimen. Historically, bedaquiline has been initiated with a relatively high loading dose (400 mg for adults), to be administered every day for the first 14 days, followed by a lower dose (200 mg for adults) to be administered three times a week for the remaining duration of treatment. Pragmatically, to allow at least 48 hours between doses, the thrice-weekly lower dose is usually administered on a Monday, Wednesday and Friday. This was the dosing schedule used in both strategies in the BEAT Tuberculosis trial in South Africa and in the BEAT-India cohort study in India. However, with the introduction of the BPaLM regimen in 2022, an alternative bedaquiline dosing schedule was introduced. Bedaquiline may be initiated at a dose of 200 mg (for adults) to be administered daily for 8 weeks, followed by a lower dose of 100 mg (for adults), which is also administered daily. This alternative once-daily dosing schedule may be simpler and more practical for some patients to administer, and may be considered for adults using the BDLLfxC regimen. Children and adolescents require weight-based dosing of bedaquiline using dispersible formulations where appropriate and accessible. Bedaquiline dose adjustment is not required for patients with mild-to-moderate renal or hepatic impairment.
Delamanid
Delamanid is administered orally twice daily, at a dose of 100 mg for adults for the first 8 weeks, followed by 200 mg once daily for the rest of the treatment. This was the dosing schedule used in both strategies in the BEAT Tuberculosis trial and in the BEAT-India cohort study. Lower doses should be used for children and adolescents, and dispersible delamanid tablet formulations should be used where appropriate and accessible. The once-daily dosing of delamanid in children and younger adolescents has not yet been studied as of 2024. Delamanid dose adjustment is not required for patients with mild-to-moderate renal impairment or in those with mild hepatic impairment.
Linezolid
Linezolid is dosed orally at 600 mg once daily for adults. This was the dosing schedule used in both the BEAT Tuberculosis trial and the BEAT-India cohort study. For children and adolescents weighing less than 46 kg, weight-based dosing is required. Younger children should receive dispersible linezolid tablet formulations (or liquid suspensions) where appropriate and accessible. Linezolid dose adjustment is not required for patients with any renal or hepatic impairment.
Levofloxacin
Levofloxacin is dosed orally at 750–1000 mg once daily for adults, depending on weight. This was the dosing schedule used in both strategies in the BEAT Tuberculosis trial. Weight-based doses should be used for children and adolescents, and dispersible levofloxacin tablet formulations should be used where appropriate and accessible, as a priority for younger children. Levofloxacin dose adjustment is required for patients with renal impairment where creatinine clearance is less than 50 mL/min. No dose adjustment is required for patients with hepatic disease.
Clofazimine
Clofazimine is dosed orally at 100 mg once daily for adults. For children and adolescents, doses should follow the latest WHO weight-based dosing table (Annex 4), using clofazimine tablet formulations that can be dispersed or 50 mg gel capsules. Clofazimine dose adjustment is not required for patients with any renal impairment or for those with mild-to-moderate hepatic impairment.
Pregnancy and postpartum
In the absence of pharmacokinetic data and specific dosing recommendations for second-line TB medications during pregnancy and postpartum, pregnant or breastfeeding women included in either strategy of the BEAT Tuberculosis trial were dosed as described above for adults, based on their total body weight. Pharmacokinetic and safety data are severely lacking in this population, and the inclusion of pregnant and breastfeeding people in TB clinical research should be prioritized.
Duration
The expected duration of the BDLLfxC, BDLLfx and BDLC regimens is 6 months (24 weeks) but this may be extended to 9 months (36 weeks) if necessary.
Although most participants on the study strategy in the BEAT Tuberculosis trial had successful treatment outcomes following 24 weeks of BDLLfxC, BDLLfx or BDLC, some participants were required to extend treatment beyond 6 months. If patients had extensive pulmonary disease, had slow clinical response to treatment or sputum culture conversion had not occurred by the fourth month (week 16) on treatment with any of the BDLLfxC regimen variations, the study protocol allowed for the full duration of the regimen to be extended by another 3 months, up to a total of 9 months (36 weeks) of treatment. A similar approach was taken in the BEAT-India study, where the BDLC regimen was extended by 12 weeks for patients with pre-XDR-TB if the week 16 sputum sample was culture positive (45). There are three main reasons for delayed culture conversion. The first is an extensive pulmonary TB disease as assessed on CXR. If possible, the CXR taken at the start of treatment should be reviewed and compared with the one taken at month 4 in this case. The second reason is non-adherence to treatment. It is necessary to check on-time pharmacy pickups and to confirm with the patient and any treatment supporter that all doses have been taken. The last reason is that there is undiagnosed resistance to one of the drug components of the regimen. This could have been present at the time of starting therapy or acquired during treatment. Where possible, a specimen should be sent for DST for bedaquiline, linezolid and delamanid.
In practice, sputum culture results usually take up to 6 weeks to be reported as negative. Therefore, bacteriological culture conversion (i.e. two consecutive cultures from samples taken on different occasions at least 7 days apart are negative) might only be confirmed after 5 or 6 months on treatment, which is when the results of samples taken at months 3 or 4 would become available. In some situations, e.g., where the full DST results are not available or where a person’s symptoms are slow to improve, clinicians may use their discretion and choose to extend treatment to nine months for patients with extensive pulmonary disease. Once a decision has been made to extend treatment beyond 6 months, further sputum culture results must be followed up, to ensure that culture conversion occurs soon after month 4 on treatment. If the month 5 and 6 culture results remain persistently positive, treatment failure should be suspected, particularly if the patient has had suboptimal adherence to treatment or shows other signs of poor clinical or radiological response to treatment.
Extending treatment because of delayed culture conversion (by month 4) is applicable mostly to individuals with bacteriologically confirmed pulmonary MDR/RR-TB. For young children diagnosed with TB based on clinical criteria or for patients with extrapulmonary TB, this extension can be applied based on clinical assessment of the treatment response, while considering other potential causes for deterioration (e.g. undiagnosed resistance, poor adherence to therapy or other clinical factors). The absence of clinical improvement or culture conversion by month 6 should prompt a comprehensive evaluation of the treatment regimen to assess potential failure.
4.2.3 Modifications of treatment
Drug toxicity
Linezolid is the least well tolerated drug within the BDLLfxC regimens owing to its toxicity profile (well described in Annexes 1 and 2); hence, many patients may be unable to continue this drug for the full 6 months of treatment. Among the 402 participants in the BEAT Tuberculosis trial in South Africa, 10% experienced severe anaemia during their treatment course (see Annex 2). Of the 202 participants in the study strategy, 159 (79%) completed the entire 6-month course of treatment without interruptions. Eighteen participants (11%) had linezolid permanently discontinued during the 6-month treatment course owing to anaemia (n=3), optic neuritis (n=6) or peripheral neuropathy (n=9). Patients initiating the BDLLfxC regimen should be supported to continue linezolid for at least the first 2 months (9 weeks) of treatment. Otherwise, an alternative regimen should be considered. Patients who experience severe AEs and have to stop linezolid beyond 2 months of treatment do not necessarily need to switch regimens, but this depends on the timing of drug withdrawal, the availability of other DST results and the patient’s clinical response to treatment. For example, if a patient with FQ-susceptible MDR/RR-TB is responding well to treatment (bacteriologically and clinically) with BDLLfx but experiences a severe linezolid-related AE after 2 months of treatment, then linezolid could be withdrawn without having to switch regimens. See Annex 2 for further details on the management of linezolid toxicity.
Severe AEs leading to withdrawal of other component drugs in the BDLLfxC regimen are rare, but permanent withdrawal of bedaquiline, delamanid or clofazimine would certainly necessitate changing to an alternative regimen. For example, if a patient’s QT interval exceeds 500 ms at any point throughout treatment, then all drugs should be withheld, and other causes of QT prolongation (electrolyte disturbances, other non-essential drugs) excluded and appropriately managed, until the QT interval is back below 500 ms. If at least four effective component drugs cannot be reintroduced without persistent QT-interval prolongation above 500 ms, then an alternative regimen must be considered.
Drug substitution
Moxifloxacin has a much stronger effect on the QT interval than levofloxacin; therefore, moxifloxacin is not considered for substitution of levofloxacin within the BDLLfxC regimens. If levofloxacin is withdrawn from a BDLLfx regimen owing to levofloxacin-related toxicity before completion of treatment, clofazimine may be used to substitute levofloxacin; the patient can then be managed as if they had FQ-resistant MDR/RR-TB and can continue treatment with the BDLC regimen instead.
Missed doses
Ideally, missing doses of all four or five drugs in the BDLLfxC regimens should be avoided; however, many patients experience occasional treatment interruption. In general, if a patient misses less than 1 week’s worth of doses over the course of their treatment, there is no need to extend treatment beyond the planned duration. However, any interruption longer than 7 consecutive or non-consecutive days (but <1 month) over the entire treatment course should be made up for by extending the treatment duration (to account for the number of missed doses). In total, 24 or 36 weeks of prescribed doses should be completed within an overall period of 7 or 10 months, respectively. This does not apply to specific medications that are interrupted owing to AEs.
Discontinuation and change to another treatment regimen
Occasionally, patients who have started the BDLLfxC regimens may have to discontinue the regimen and change to a different treatment regimen, or vice versa; for example, because of the risk of potential AEs of component drugs within the regimen (e.g. patients becoming pregnant while receiving pretomanid within BPaL or BPaLM); following receipt of DST results indicating resistance to component drugs within the regimen; or simply because clinicians and their patients may have reason to prefer a different regimen for which the participant is eligible. Examples of such situations are outlined below:
- Patients receiving treatment with BDLLfx, BDLC or BDLLfxC will have to change to a different regimen if bedaquiline or delamanid (or clofazimine in the case of FQ-resistant MDR/RR-TB) needs to be withdrawn at any point in treatment, or if linezolid use is limited by toxicity within the first 2 months of treatment. Drug withdrawal may be due to toxicity or intolerance, sustained interruption in drug supply or evidence of resistance to any of the drugs in the regimen (bedaquiline, delamanid, linezolid or clofazimine).
- Patients will have to change to a more appropriate regimen if the BDLLfxC regimens appear to be failing, as evidenced by poor clinical, bacteriological or radiological response (or further deterioration), or sustained reconversion of negative cultures to positive at any point in treatment.
4.2.4 Key subgroups
Extensive pulmonary TB and resistance to fluoroquinolones
Extensive pulmonary TB is defined as the presence of bilateral cavitary disease or extensive parenchymal damage on CXR. Patients with extensive pulmonary MDR/RR-TB disease were included in both the study strategy and the control strategy (but treated for 9 months) in the BEAT Tuberculosis trial in South Africa, and most of the participants in the BEAT-India study in India had extensive bilateral disease (45); despite this, both studies reported high rates of treatment success with the 6-month study regimens. In some situations (e.g. where the full DST results are not available or where a person’s symptoms are slow to improve), clinicians may use their discretion and choose to extend treatment to 9 months for patients with extensive pulmonary disease.
Patients with DR-TB who have extensive disease face greater challenges in treatment across all regimens and are at a higher risk of unfavourable outcomes and amplification of drug resistance. This risk is further elevated in those infected with pathogens that have advanced resistance profiles, particularly with additional resistance to fluoroquinolones. These patients may experience slightly worse outcomes compared to those without pre-XDR and develop resistance to regimen components. The evidence from the BEAT-TB trial is insufficient to draw definitive conclusions due to the small sample when stratified by fluoroquinolone resistance and related imprecision. Nonetheless, clinicians should remain vigilant and closely monitor the treatment response in this group of patients and promptly take relevant actions (extending the duration of treatment or changing the regimen).
Extrapulmonary TB
The BEAT Tuberculosis trial in South Africa included patients with non-severe or uncomplicated extrapulmonary TB if they also had pulmonary disease; however, it excluded patients with tuberculous meningitis, miliary TB, and pericardial and osteoarticular disease. Therefore, the BDLLfxC regimens should not be offered to patients with severe or complicated extrapulmonary MDR/RR-TB disease involving the CNS, or osteoarticular and disseminated forms of TB because these patients require treatment with a regimen of longer duration.
People living with HIV
The 6-month BDLLfxC regimen was evaluated in a high HIV prevalence setting; 50% of participants enrolled on the BEAT Tuberculosis trial in South Africa were PLHIV. There is no reason to believe that this regimen would perform differently in people who initiate appropriate ART in a timely manner and are adequately supported to adhere to treatment for both diseases. In BEAT Tuberculosis 82.9% of PLHIV had a favorable treatment outcome with the six-month regimen compared with 89.7% of participants without HIV. As safer and more tolerable medications become more widely available, DDIs and overlapping toxicities are becoming less common. In settings where zidovudine is still used, co-administration of this drug with linezolid increases the risk of myelosuppression and thus should be avoided. Non-nucleoside reverse transcriptase inhibitors were prohibited in the BEAT Tuberculosis trial in South Africa because of DDIs with bedaquiline (efavirenz significantly reduces the concentration of bedaquiline, which may reduce efficacy of the drug). In addition, co-administration of efavirenz with delamanid increases the risk of neuropsychiatric adverse effects. Therefore, alternative ARVs should be used with the BDLLfxC regimens – in the BEAT Tuberculosis trial, ART for PLHIV included either dolutegravir or a protease inhibitor. There are no overlapping toxicities or DDIs between dolutegravir and any of the component drugs in the BDLLfxC regimens, but interactions with other integrase inhibitors and newer ARV drugs have not yet been studied.
Pregnant and breastfeeding women
Data on dosing and safety to support the optimal use of second-line anti-TB medicines during pregnancy remain sparse, highlighting the need for wider inclusion of pregnant and breastfeeding women in TB research. In general, the benefits (to both mother and fetus or infant) of providing effective MDR/RR-TB treatment to the mother far outweigh the potential risks posed by these medications to the fetus in-utero or to the breastfeeding infant.
The BEAT Tuberculosis trial allowed for the enrolment or retention of pregnant and breastfeeding women on both the study strategy and the control strategy. Among the 10 pregnant women enrolled in the BEAT Tuberculosis trial overall, four were randomized to the study strategy with the 6-month BDLLfxC regimens. All pregnancies in this trial resulted in singleton live births, with one preterm delivery. No drug-related and no severe AEs were reported among the women or their infants; however, one of the four women randomized to the study strategy experienced a relapse of TB following completion of treatment. Although numbers are too small to draw any definitive conclusions, these findings highlight the need to support and optimize adherence through treatment and provide post-treatment follow-up for at least 6 months beyond treatment completion, to rapidly identify cases of relapse in pregnant and postpartum women.
Care providers for pregnant women with MDR/RR-TB must also pay particular attention to the seamless continuity of care between antenatal and TB services, which are not integrated in most TB-endemic settings. Good communication with the patient and among various providers is essential to avoid unnecessary treatment modifications to the BDLLfxC regimens, and to reduce stigma and potential LTFU from treatment. Patients require considerable adherence support and monitoring of proper administration of MDR/RR-TB treatment and other medications in the early postpartum period, when chronic medication might not be prioritized, to ensure successful maternal treatment outcomes and minimal risk of TB transmission from mother to infant.
Children and adolescents
The BEAT Tuberculosis trial in South Africa allowed enrolment of children and adolescents aged above 6 years to either treatment strategy. In total, 30 younger patients aged 8–17 years were enrolled on the trial. Thirteen children were randomized to the study strategy group and all had bacteriologically confirmed MDR/RR-TB disease, of which 24% were FQ-resistant.
Most of the children and adolescents enrolled on the BEAT Tuberculosis trial were aged above 12 years. Bacteriological confirmation of MDR/RR-TB disease in children aged below 12 years often signifies severe disease. The high rate of treatment success among children and adolescents in this trial is encouraging; it supports the efficacy of this regimen for children of all ages with severe and non-severe pulmonary MDR/RR-TB disease. In the absence of bacteriological confirmation of disease in younger children, the decision to treat for MDR/RR-TB relies on clinical signs and symptoms of TB disease, radiological findings, and history of significant exposure to a person with microbiologically confirmed MDR/RR-TB. In these cases, treatment decisions and regimen choice should be based on the drug-resistance pattern of the isolate obtained from the index case.
Despite the small numbers of children included in the BEAT Tuberculosis trial, the evidence supporting the efficacy of this regimen in adults and adolescents may be extrapolated to children, provided the implementation considerations are followed. Therefore, children of all ages, with either severe or non-severe pulmonary disease, as well as those with uncomplicated extrapulmonary disease (peripheral lymph nodes or isolated mediastinal mass without airways compression) are likely to benefit from the 6-month BDLLfxC regimen.
All the component drugs in the BDLLfxC regimens have already been used in various combinations to treat MDR/RR-TB in children, and drug dosages have been established (and continue to be refined) for children of all ages. Child-friendly, dispersible formulations exist for all the component drugs in the BDLLfxC regimens and are strongly preferred by children and their caregivers. TB programmes must prioritize procuring and providing these formulations for children with MDR/RR-TB. Although child-friendly formulations are the priority, the lack of these is not a barrier to providing children access to the BDLLfxC and other MDR/RR-TB regimens. Bedaquiline and delamanid tablets can be crushed and mixed in water for easier administration to young children without compromising the bioavailability of the drugs (in comparison to swallowing tablets whole), and linezolid is also available as a suspension for easier administration in children who are unable to swallow tablets.
Although children tend to tolerate treatment better than adults, the AEs associated with second-line anti-TB drugs remain significant for children, especially as they may be unable to adequately verbalize or describe the symptoms they experience. This underscores why shorter regimens and those involving fewer drugs, such as the BDLLfxC regimen, are particularly favored for children. However, it remains essential to stay vigilant about the potential adverse effects of these medications and to monitor children closely for any such reactions.
Peripheral neuropathy associated with linezolid is less commonly reported by children than adults, and severe myelosuppression may occur relatively quickly without any obvious symptoms, highlighting the importance of regular blood monitoring. Among all 30 child and adolescent participants enrolled on the BEAT Tuberculosis trial, there were three severe AEs related to linezolid (anaemia, peripheral neuropathy and optic neuritis). Compared with adults, children appear to experience more neuropsychiatric AEs related to delamanid; therefore, clinical monitoring should include regular assessment for persistent nightmares or night terrors with sleep disturbances (46). In the BEAT Tuberculosis trial, none of the children enrolled in the study strategy discontinued delamanid due to neuropsychiatric AEs.
4.2.5 Implementation considerations
DST results
Although DST for FQ is strongly encouraged, initiation of MDR/RR-TB treatment is sometimes based on just a rifampicin-resistant DST result, and it may be the case that no further information on DST for other drugs is ever obtained. This may be due to limited diagnostics capacity, sample leakages, unsuccessful test results, or inability to perform DST on negative or contaminated TB cultures. In such cases, patients who fulfil other eligibility criteria for the 6-month regimens may initiate BDLLfxC and continue treatment with all five drugs. With increasing access to novel treatment regimens and the recent emergence of bedaquiline-resistant strains of MDR/RR-TB in some high TB burden settings (47–49), wider access to more rapid diagnostics for timely detection of resistance to component drugs within the 6-month regimens is becoming more important. The 6-month BDLLfxC regimen is not considered effective and is not recommended for MDR/RR-TB with confirmed resistance to bedaquiline, linezolid, delamanid or clofazimine. Given the unavailability of DST in several settings or the considerable delay in obtaining DST results for these drugs, alongside the urgent need to initiate treatment early to reduce ongoing MDR/RR-TB transmission, NTPs are encouraged to use the results of surveillance testing to identify risk factors or groups of patients at increased risk of harbouring bedaquiline-resistant strains, and provide a framework for appropriate management of such patients.
History of exposure to component drugs
A thorough history of prior anti-TB treatment, as well as extent of exposure to people with confirmed MDR/RR-TB (and their full drug susceptibility profile), is necessary to assess an individual’s risk of potential exposure to, or acquisition of, MDR/RR-TB with additional drug resistance. This is especially important in people initiating MDR/RR-TB treatment without confirmation of the full drug susceptibility profile of the infecting Mtb strain; for example, in people diagnosed with RR-TB and no further DST results, and in children with a clinical diagnosis of MDR/RR-TB disease. Prior exposure to bedaquiline, delamanid, linezolid or clofazimine for more than 1 month, particularly in a recent treatment episode that was interrupted or failed, signifies a potentially increased risk of that person harbouring more extensively resistant MDR/RR-TB strains.
Management of anaemia and myelosuppression
The risk of myelosuppression associated with even relatively short exposures to linezolid is a significant concern in patients with anaemia at the time of MDR/RR-TB treatment initiation. Patients with TB often have anaemia of chronic disease, and iron deficiency is common in children with malnutrition. Pregnant women with TB are likely to be anaemic (for multiple reasons) before starting treatment. Therefore, pretreatment assessment of Hb, neutrophils and platelets is crucial in patients considering treatment with linezolid-based regimens. Children with a low baseline Hb are at risk of developing severe linezolid-induced haematological toxicity (50). The 6-month BDLLfxC regimens may not be suitable for patients with a pretreatment serum Hb level below 8 g/dL. Blood transfusions can be beneficial in quickly correcting Hb levels before starting MDR/RR-TB treatment, enabling the initiation of linezolid. The acceptability and availability of blood transfusion in various settings must be taken into consideration and balanced alongside the risks and benefits of treating with shorter, linezolid-containing regimens. Treatment with an effective MDR/RR-TB regimen (even including linezolid) usually leads to improvement or resolution of anaemia once the disease is properly treated and the patient’s appetite improves. Regular blood tests are necessary to monitor for linezolid-induced myelotoxicity; this may be challenging, especially in younger children, and health care providers could consider alternative feasible options for monitoring (e.g. rapid Hb tests on heelstick samples).
Blood transfusions during treatment may or may not be helpful in situations where Hb drops significantly owing to linezolid toxicity. The effects of a blood transfusion to correct linezolid-induced anaemia are often transient if exposure to linezolid is ongoing; however, a transfusion might assist a patient in completing at least the first 2 months of linezolid and may also help to avoid the person having to switch to a longer regimen. Aside from blood transfusions, it may be possible to manage linezolid-induced anaemia by pausing the drug for short periods (7–14 days) or reducing the dose of linezolid (although this is not recommended in patients weighing over 50 kg). In the BEAT-India study, where adults with pre-XDR-TB were treated with the 6-month BDLC regimen, patients were initiated on linezolid at a dose of 600 mg once daily; 100 (60%) of the 165 patients in this cohort had a BMI of less than 18.5 kg/m². The drug dose was reduced to 300 mg daily in 45 patients experiencing linezolid-induced severe AEs, following a temporary pause in linezolid dosing, and 40 of these 45 patients went on be cured of their disease. The approach to linezolid toxicity management in the BEAT Tuberculosis trial in South Africa, where 10% of participants receiving linezolid in the study strategy experienced severe anaemia (Hb <8 g/dL) (51), was to withhold linezolid for a short period, rechallenge the drug at the same dose and transfuse the patient. Reduced doses of linezolid (<600 mg for adults) were not used in the BEAT Tuberculosis trial.
Owing to the morbidity associated with severe neutropenia and thrombocytopenia, the 6-month BDLLfxC regimen is also not suitable in patients with neutrophil levels of less than 750/mm3 or platelets below 150 000/mm3 before starting treatment. However, because these parameters may change quickly, it is advisable to repeat them before making a decision on withholding BDLLfxC.
QT-interval prolongation
The risk of QT-interval prolongation increases with exposure to an increasing number of QT-prolonging drugs in the regimen; hence, the risk is higher for patients receiving regimens containing bedaquiline and delamanid as well as clofazimine. Among these medicines, the effect of clofazimine appears to be the strongest, with a QT-interval change from baseline of almost 30 ms at currently recommended doses. The QT-prolonging effects of these drugs are likely to be additive in adults with normal baseline QT-interval values, and although the combination of these three drugs may not necessarily result in clinically significant QT-interval prolongation beyond 500 ms, patients receiving the BDLC or BDLLfxC regimens will have less leeway for further QT-interval variation. Therefore, patients with a QT interval closer to 500 ms at baseline, or those with other risk factors for QT-interval prolongation (e.g. thyroid dysfunction, electrolyte disturbances, diabetes or concomitant use of other QT-prolonging medications) should be informed of their potentially higher risk of clinically significant QT-interval prolongation; such patients should receive closer ECG monitoring and have reversible causes of QT-interval prolongation corrected as soon as possible. Results from the BEAT-India study in India are encouraging; 165 adult participants received the 6-month BDLC regimen but none were reported to have QT-interval prolongation above 500 ms, despite some participants receiving higher (200 mg) daily doses of clofazimine (45). The incidence of severe QT-interval prolongation also appears to be lower in children than in adults, possibly because of a combination of fewer cardiac comorbidities and exposure to relatively low anti-TB drug concentrations.
Treatment interruption
In the BEAT Tuberculosis trial in South Africa, patients on the study strategy whose treatment was interrupted for more than 4 consecutive weeks and who did not restart the 6-month BDLLfxC regimens were considered to be lost to follow-up. Treatment interruption over such a period increases the risk of selective drug pressure on MDR/RR-TB strains owing to the long half-life of bedaquiline (and, to some extent, clofazimine). Patients returning to care after 4 or more weeks of treatment interruption are likely to be at higher risk of harbouring further drug-resistant MDR/RR-TB strains, in which case the 6-month BDLLfxC regimen is inadequate. The risk of treatment failure is highest in patients without any evidence of sputum culture conversion before the period of interruption, and these patients require a different treatment regimen when they return to care.
Care and support
To ensure optimal efficacy of the BDLLfxC regimen, medications should preferably be administered with food, especially as this increases the bioavailability of some drugs (bedaquiline, delamanid, clofazimine). However, where possible, calcium-containing foods and supplements should be avoided for 2 hours before and after administration of levofloxacin because calcium binding renders the drug ineffective. People receiving the BDLLfxC regimen must be supported using a person-centred approach to ensure full adherence to treatment. The principles of person-centred care and support for people with TB are outlined in Chapter 3 of this handbook. Specific adherence challenges should be identified early in treatment, and locally feasible strategies employed to support patients at home and in their communities to complete the full duration of treatment; patients with such challenges should also be given information on appropriate treatment administration options. Child-friendly formulations of all drugs in the BDLLfxC regimen are available and should be procured and provided to all children with MDR/RR-TB.
Early counselling for patients and their treatment supporters should include information on potential side-effects that patients may experience with the BDLLfxC drugs, particularly the neuropsychiatric effects of delamanid in children (which may be alarming for parents) as well as the skin discolouration with clofazimine (which parents may not necessarily be expecting in infants exposed in-utero or while breastfeeding). In addition, patients (and their caregivers) initiating the 6-month regimens should be counselled before or early in treatment about the consequences of interrupting the 6-month BDLLfxC regimens, because subsequent treatment options when they return to care are likely to be more challenging in terms of toxicity and duration of treatment.
Cost–effectiveness analysis
Cost–effectiveness studies for the 6-month BPaL regimen and for the BDLC regimen in India (BEAT-India) demonstrated both regimens to be cost-saving compared with the longer MDR/RR-TB regimens, largely by virtue of the shorter duration of treatment with expensive medications. Although these findings may be roughly extrapolated to the 6-month BDLLfxC regimen, no cost–effectiveness analysis has been carried out specifically for the BDLLfxC regimens in South Africa.
14 Dosages of the other drugs are given in Table 2.4.1.
15 The permitted antiretroviral treatments were nevirapine in combination with tenofovir/abacavir or emtricitabine/lamivudine; lopinavir/ritonavir in combination with tenofovir/abacavir or emtricitabine/lamivudine; and integrase inhibitor in combination with tenofovir/abacavir or emtricitabine/lamivudine. For patients on efavirenz, therapy could be changed to rilpivirine with tenofovir/abacavir or emtricitabine/lamivudine.
16 Prices use the lowest price available from Global Drug Facility (GDF), effective from 1 January 2025, and are subject to change.
17 Extensive (advanced) pulmonary TB disease is the presence of bilateral cavitary disease or extensive parenchymal damage on chest radiography. In children and young adolescents aged below 15 years, advanced disease is usually defined by the presence of cavities or bilateral disease on CXR.
18 Severe extrapulmonary TB is the presence of miliary TB and TB meningitis, osteoarticular and pericardial TB. In children and young adolescents aged below 15 years, extrapulmonary forms of disease other than lymphadenopathy (peripheral nodes or isolated mediastinal mass without airway compression) are considered to be severe.
