
 Retour
RetourA3.1 Background
Pharmacovigilance is defined by the World Health Organization (WHO) as “the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem”. It is a fundamental public health surveillance activity designed to inform the management of patient safety measures in health care. Pharmacovigilance is a facet of programme monitoring, and is similar to the way many countries operate routine surveillance of tuberculosis (TB) drug resistance based on diagnostic testing.
Patients can be better served if monitoring of drug safety is implemented in tandem with management of adverse events (AEs) and adverse drug reactions (ADRs). Many of the second-line anti-TB drugs are more likely to cause toxic reactions in patients than first-line drugs, making pharmacovigilance more important in programmatic management of drug-resistant TB (PMDT). By recording the occurrence of ADRs for patients on treatment, many programmes are already collecting basic data inherent to pharmacovigilance. However, the collection of such data and the measurement of indicators on pharmacovigilance are not part of the standard parameters used in the monitoring of TB patients on treatment. Consequently, in most programmes, the nature and frequency of harm caused by the drugs themselves are poorly profiled, and they can only be inferred indirectly, from interruption or failure of treatment. As programmes start to incorporate newly released drugs into treatment regimens, WHO recommends that capacity to undertake pharmacovigilance also be improved, because this is fundamental in ensuring the safety of patients and the updating of patient safety standards, drug-safety profiles and TB treatment guidelines.
In November 2014, a WHO workshop with broad representation from stakeholders and experts was held in Viet Nam, to define methods for active surveillance of drug-safety concerns in TB programmes (1). To improve understanding and arrive at a broad consensus on ways to address patient safety, the WHO Global TB Programme (WHO/GTB) convened a consultation meeting in Geneva, Switzerland, for key technical partners on 28–29 July 2015. The technical partners discussed essential requirements for the implementation of active pharmacovigilance and proper management of AEs and ADRs, which is one of the conditions included in the WHO interim policies on the use of new anti-TB medicines or novel multidrug-resistant TB (MDR-TB) regimens. The consensus reached during this meeting and in subsequent discussions is presented in the framework for the implementation of active TB drug-safety monitoring and management (aDSM) (2). The framework outlines the agreed essential requirements for aDSM in patients on treatment for drug-resistant TB (DR-TB), and proposes key terms that were adapted to the specific context of TB drug-safety monitoring. This section provides advice on implementing the WHO policy on aDSM for the treatment of MDR-TB. TB practitioners, health officials, planners, public health teams, drug regulatory authorities and others should become familiar with other publications relating to the subject (3, 4).
A3.2 Definitions used in aDSM⁵
Active TB drug-safety monitoring and management (aDSM) is the active and systematic clinical and laboratory assessment of patients on treatment with new anti-TB drugs, novel MDR-TB regimens or extensively drug-resistant TB (XDR-TB) regimens to detect, manage and report suspected or confirmed drug toxicities. Although all detected AEs need to be managed, the core package of aDSM requires the reporting of serious AEs (SAEs) only. MDR-TB and XDR-TB treatment sites with additional resources may also monitor other AEs that are of clinical significance or of special interest to the programme, as part of comprehensive aDSM.
Adverse drug reaction (ADR) is a response to a TB medicine that is noxious and unintended, and that occurs at doses normally used in humans.
Adverse event (AE) is any untoward medical occurrence that may present in a TB patient during treatment with a pharmaceutical product, but does not necessarily have a causal relationship with this treatment.
Serious adverse event (SAE) is an AE that leads to death or a life-threatening experience, hospitalization or prolongation of hospitalization, persistent or significant disability, or a congenital anomaly. The definition includes SAEs that do not immediately result in one of these outcomes but may require an intervention to prevent it from happening. SAEs may require a drastic intervention, such as termination of the drug suspected of having caused the event.
Adverse event of special interest is an AE documented to have occurred during clinical trials and for which the monitoring programme is specifically sensitized to report, regardless of its seriousness, severity or causal relationship to the TB treatment. The centres that offer intermediate and advanced packages of aDSM will include all AEs of special interest in their reporting.
Adverse event of clinical significance is an AE that is serious, is of special interest, leads to a discontinuation or change in the treatment, or is otherwise judged as being clinically significant by the clinician. The centres that offer the advanced package of aDSM will include all AEs of clinical significance in their reporting.
Adverse event leading to treatment discontinuation or change in drug dosage is an event that leads a clinician to stop, interrupt temporarily or change the dosage of one or more drugs, regardless of its seriousness, severity or causal relationship to the TB treatment.
Causal relationship is a relationship between an exposure (A) and an event (B) in which A precedes and causes B. This may refer to the causal association between an exposure to a TB medicine and the occurrence of an adverse reaction.
Causality assessment is the evaluation of the likelihood that a TB medicine was the causative agent of an observed adverse reaction.
Drug-safety profile is a description of the benefits, risks and toxicity of a given TB drug or regimen, specifying any known or likely safety concerns, contraindications, cautions, preventive measures and other features that the user should be aware of to protect the health of a TB patient.
Sentinel sites are centres that, in addition to the core package of aDSM, also undertake intermediate or advanced levels of drug-safety monitoring.
Signal is reported information on a possible causal relationship between an AE and a TB medicine, the relationship being unknown or incompletely documented previously or representing a new aspect of a known association. It covers information arising from one or multiple sources that is judged to be of sufficient likelihood to justify verification (5).
A3.3 What to monitor for aDSM
WHO recommends aDSM for all MDR/RR-TB patients treated with new medicines (e.g. bedaquiline, delamanid or pretomanid), repurposed medicines (e.g. linezolid, clofazimine) or novel regimens (e.g. 6-month BPaLM/BPaL regimen, 9-month all-oral regimen or other regimens that include multiple new and repurposed drugs). The appropriate and timely management of all AEs and ADRs is an integral component of aDSM and patient care.
A priority for monitoring is the detection of SAEs that lead to hospitalization or prolongation of hospitalization, a persistent significant disability, a congenital anomaly, a life-threatening condition or death. It is necessary to report SAEs as per the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guideline definition (6). All deaths are to be reported and as much relevant information as possible on the cause of death should be consistently collected. This may require recovering information from vital registration coding. Reporting of AEs and other events (e.g. pregnancy and lactation exposure) may be required, primarily based on what is known about the safety profile of the new agent and also for other possible harms that have not yet been described.
All reasonable measures are thus needed to ensure that patient safety is monitored alongside the effectiveness of treatment. In this situation, spontaneous reporting is not expected to represent an appropriate level of care, and active and cohort-based drug-safety monitoring approaches are considered necessary to improve early and systematic detection, and management of harms. It is also important to collect safety data accurately, to ensure that any AE is properly investigated and no hasty conclusions about the causative medicine are drawn. A cohort approach is essential to avoid bias in the selection of patients or in the measurement of events; it is also the best way to infer the potential association of an event with the given exposure, and it provides denominators and baseline data for analysis. Many TB practitioners are unfamiliar with the concept of “cohort event monitoring” and other conventional terminology of pharmacovigilance, making it difficult for them to follow recent recommendations for introduction of a drug-safety monitoring component in PMDT programmes. The aDSM approach therefore outlines the agreed “essential requirements for active drug-safety monitoring and management in patients on treatment for drug-resistant TB”. It proposes key terms that have been adapted to the specific context of active TB drug-safety monitoring. This adaptation should help the TB community to speak a common language while implementing the required drug-safety activities.
The recording and reporting of aDSM primarily target SAEs as a core requirement. PMDT sites with additional resources may also monitor other AEs that are of clinical significance or of special interest to the PMDT programme as part of an extended aDSM approach.
aDSM is important when patients are treated with a medicine for which the drug-safety profile is not yet complete. This does not depend on the number of patients enrolled. Monitoring needs to be closely associated with early action to prevent and manage any serious consequences to the individual patient. A national programme should also strive to capture data in the private sector and through public–private partnerships.
aDSM is intended to pick up not only known reactions associated with a drug but also any unexpected effect of treatment (some of these may actually be beneficial to the patient). For aDSM, a non-severe event may be the early manifestation of a more consequential process (e.g. a dose-dependent effect).
Where it is feasible for the programme, such events should be captured on data collection forms. To reduce the workload, entering of this information into the aDSM database should be optional.
A3.4 Objectives of aDSM
The overall objectives of aDSM are to reduce risks from drug-related harms in patients on second-line treatment for DR-TB and to generate standardized aDSM data to inform future policy updates on the use of such medicines. To achieve these objectives, the aDSM includes four essential activities:
- Patients targeted for aDSM should undergo active and systematic clinical and laboratory assessment during treatment to detect drug toxicity and AEs.
- All AEs detected should be managed in a timely manner, to deliver the best possible patient care (as described in Annex 2).
- Standardized data should be systematically collected and reported for any SAE detected:⁶ these data will eventually be used to characterize the types of SAEs, assess the safety of treatment and inform future policy on the use of these medicines.
- Improving the evidence base for global policy on new and repurposed medicines.
For the evidence base to improve, AE data collected by national authorities need to be shared to permit global monitoring and data pooling. This approach is also useful to detect previously unrecognized or poorly documented AEs. A global database for active TB drug-safety monitoring and management has been launched by WHO and the Special Programme for Research and Training in Tropical Diseases (TDR). The aDSM website provides details on how countries can submit data. Close coordination of aDSM activities with the main pharmacovigilance structures at the country level is essential to avoid overlap and duplication. Any future recommendation of WHO on off-label use of new anti-TB medicines (e.g. delamanid or bedaquiline) for more than 6 months will depend on the availability of good-quality safety data – proper implementation of aDSM is paramount for such data.
A3.5 Levels of monitoring in aDSM
There are three levels of monitoring in aDSM:
- core package – which requires monitoring for and reporting of all SAEs;
- intermediate package – which includes SAEs as well as AEs of special interest; and
- advanced package – which includes all AEs of clinical significance.
All PMDT sites treating eligible patients with new anti-TB drugs or novel MDR-TB regimens, or treating patients for XDR-TB, require the core package. These treatment centres should, as a minimum, also take part in spontaneous reporting of ADRs, as required by local regulations. Expansion of aDSM should be implemented in a phased approach as and when resources permit.
All SAEs detected should be reported to the national authority responsible for pharmacovigilance according to individual country requirements (including time limits for reporting) and should be regularly assessed for causality.
A3.6 Implementing aDSM
The implementation of aDSM will require the following eight essential steps:
- Create a national coordinating mechanism for aDSM.
- Develop a plan for aDSM.
- Define management and supervisory roles and responsibilities.
- Create standard materials for data collection.
- Train staff on the collection of data.
- Define schedules and routes for data collection and reporting.
- Consolidate aDSM data electronically.
- Develop (or use existing) capacity for signal detection and causality assessment.
Ideally, all eight steps should be in place before patients are enrolled on treatment with new drugs, novel MDR-TB regimens or XDR-TB treatment. Where this is not feasible, Step 4 (Create standard materials for data collection) and Step 5 (Train staff on the collection of data) are considered essential ahead of any patient enrolment.
A fully functional aDSM is not required at the time of ordering drugs or starting patients on treatment. However, certain key elements need to be in place so that essential safety data are collected for all patients as soon as they are started on a new drug or new regimen. The capacity for aDSM can then be built over the following months.
The aDSM plan would clearly define the activities and standard operating procedures (SOPs), including the plan for data collection, reporting of indicators, analysis and communication. The final document would be incorporated within the national TB or PMDT guidelines. Local or international experts in drug safety as well as the national pharmacovigilance centre (if functional) should be engaged.
Some of the data collection tools for aDSM are separate from those used for routine PMDT monitoring; nevertheless, the process could be integrated with other cohort-based monitoring for bacteriological response and outcomes that have been a standard feature of the PMDT component of TB control programmes for several years (see Section 2 of the guidelines and the corresponding annexes). WHO is working closely with partners towards further integration of aDSM within routine PMDT monitoring.
In the core package of aDSM, clinical and laboratory test records at baseline and during regular reviews (e.g. monthly intervals) would be integrated with an expanded version of the programmatic MDR-TB (second-line TB) treatment card.
Before enrolling any patients, staff at the different levels of health services would be informed and trained on the use of new anti-TB drugs or novel regimens (including instruction on the completion of aDSM forms). This activity should be completed ahead of any patient enrolment, to ensure timely identification of AEs that need to be managed, and proper and complete collection of information.
All AEs detected during routine clinical patient care should lead to an appropriate and timely management response to limit potential harms to the patient. In terms of monitoring, the minimum requirement for aDSM is that all SAEs be registered and reported, regardless of their severity or whether they were caused by any of the medicines to which the patient was exposed.
Some centres with sufficient resources may be designated as “sentinel sites” and undertake monitoring additional to that required by the core package of aDSM (e.g. the reporting of AEs of special interest or AEs of clinical significance, as described above). In many countries, the law mandates the reporting of ADRs to the national pharmacovigilance centre. In all public and private health services, TB practitioners should comply with the national legal requirements for such reporting.
A standard form (in paper or electronic format) will need to be developed to alert the programme when any SAE occurs; the content of the form should be similar to that used by the national pharmacovigilance centre for spontaneous reporting. The creation of an electronic database – or preferably the adaptation of an existing TB patient database to accommodate the additional data fields required – is an important step in aDSM implementation. It will ensure the standardization and safekeeping of data. If data are collected on paper forms, these need to be entered regularly into the electronic database. The management of data in electronic format is indispensable for facilitating data sharing and data analysis, and for generating indicators.
Measures would be taken to avoid duplication of work by revising existing databases, ensuring interoperability of data management systems, consulting with local pharmacovigilance authorities and granting access rights to users for different data as needed (Fig. A3.1). The roles and responsibilities for data management and analysis would be specified in the aDSM plan, to avoid the creation of parallel systems of ADR reporting and to make use of the best possible expertise on drug safety in the country.
Fig. A3.1 outlines the main lead responsibilities for the different components of aDSM; it could be useful in assigning complementary functions and associated funding needs. This construct is subject to adjustment based on local circumstances; for example, if the national pharmacovigilance centre has limited capacity for running an aDSM project in the country, it may be agreed that the national TB programme (NTP) or a technical agency will lead certain functions. Technical agencies could, for instance, catalyse the establishment of a committee or the protocol, organize training or provide technical assistance. Donors could have a role in supporting grant proposals for pharmacovigilance and facilitating the process for accessing the resources at country level.
In addition to the identification of signals and causality assessment, indicators will be useful for assessing the coverage of aDSM activities and summarizing the overall AE experience of monitored patients. For these purposes, Table A3.1 presents the indicators and Table A3.2 a “drug-safety profile” (1). The essential laboratory tests and examinations that need to be conducted will be determined by the programme protocol.
Development of a schedule for screening of AEs and for laboratory, clinical and radiological testing is also recommended. Both the list of data elements and the frequency of testing would be validated and customized based on local needs before they are integrated in the programme’s aDSM protocol.
Fig. A3.1. Generic model of aDSM within drug-safety structures at the national level
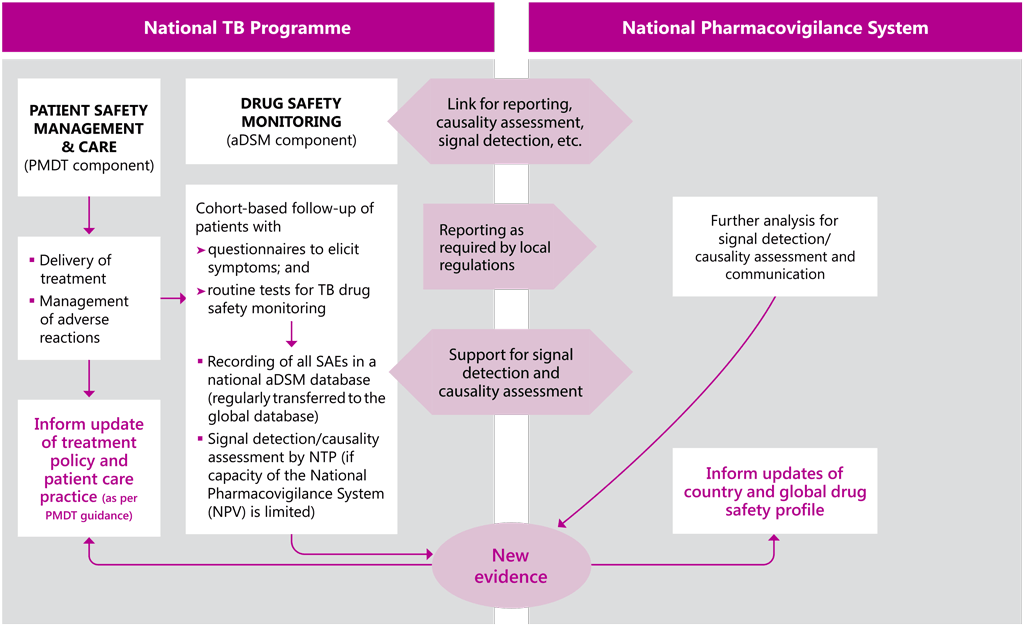
aDSM: active TB drug-safety monitoring and management; NPV: national pharmacovigilance centre; NTP: national TB programme; PMDT: programmatic management of drug-resistant TB; SAE: serious adverse event; TB: tuberculosis.
Source: WHO (2014) (7).
A3.7 Roles, responsibilities and support for the implementation of aDSM
Responsibility for the coordination of aDSM at the national level should be assigned to an existing TB expert body, such as the MDR-TB committee (or consilium) or the technical working group on new drugs. These committees should primarily have scientific and clinical expertise for MDR-TB care and drug-safety monitoring, but could also include expertise important for coordination and advocacy (e.g. funding, communication and patient representation). Until such a group is tasked with this role, the NTP needs to assign someone to coordinate the necessary aDSM activities and ensure that the key steps mentioned above are in place before patient enrolment.
The ultimate purpose of systematic data collection within aDSM is to enable causality assessment for SAEs, determine the frequency (rates) of SAEs and detect signals. Physicians skilled in MDR-TB management already attempt to assess relationships between drugs and ADRs and take appropriate clinical action. However, formal causality assessment is a separate process that requires involvement of other experts. In several countries, the capacity of national pharmacovigilance centres to conduct formal causality assessment is limited, but where such capacity exists it should be used.
NTP staff need to acquire the skills to undertake essential activities related to aDSM. This is a long-term goal but needs to be started as part of the plan to introduce new anti-TB drugs and novel MDR-TB regimens. To carry out such capacity-building, the NTP should seek local or international expertise in causality assessment; WHO is also working with partners to accelerate these efforts.
The implementation of aDSM at the NTP level will be greatly facilitated by familiarity with the concept of cohort-based follow-up of patients, which is the foundation for the monitoring and evaluation of TB and MDR-TB treatment programmes. The testing schedules used in these projects have largely followed those generally recommended when second-line TB drugs are used.
Experience from observational studies of shorter regimens for MDR-TB has shown that active drug-safety monitoring can be implemented within programmes if dedicated funding is provided. Most of the additional resources are needed to undertake clinical testing (e.g. electrocardiography and audiometry) and laboratory analyses, and to collect drug-safety data.
Once the right skills have been acquired and links have been established with appropriate experts in drug safety, it is envisaged that causality assessment and signal detection could be organized within the PMDT programme, with appropriate capacity-building and support from drug-safety experts (if such capacity is missing at the national pharmacovigilance system). More work is needed to quantify the costs of aDSM, and these will eventually be reflected in the tools that will be provided to help users with budgeting.
Clinicians treating patients with second-line anti-TB drugs are usually familiar with clinical monitoring for AEs; however, this knowledge may not be available to many other health care workers within the programme. The monitoring component of aDSM is also likely to be novel to many health care workers. WHO/GTB and technical partners will be supporting NTPs to build such capacity and to integrate aDSM into routine PMDT monitoring.
Table A3.1. Programmatic indicators for aDSM
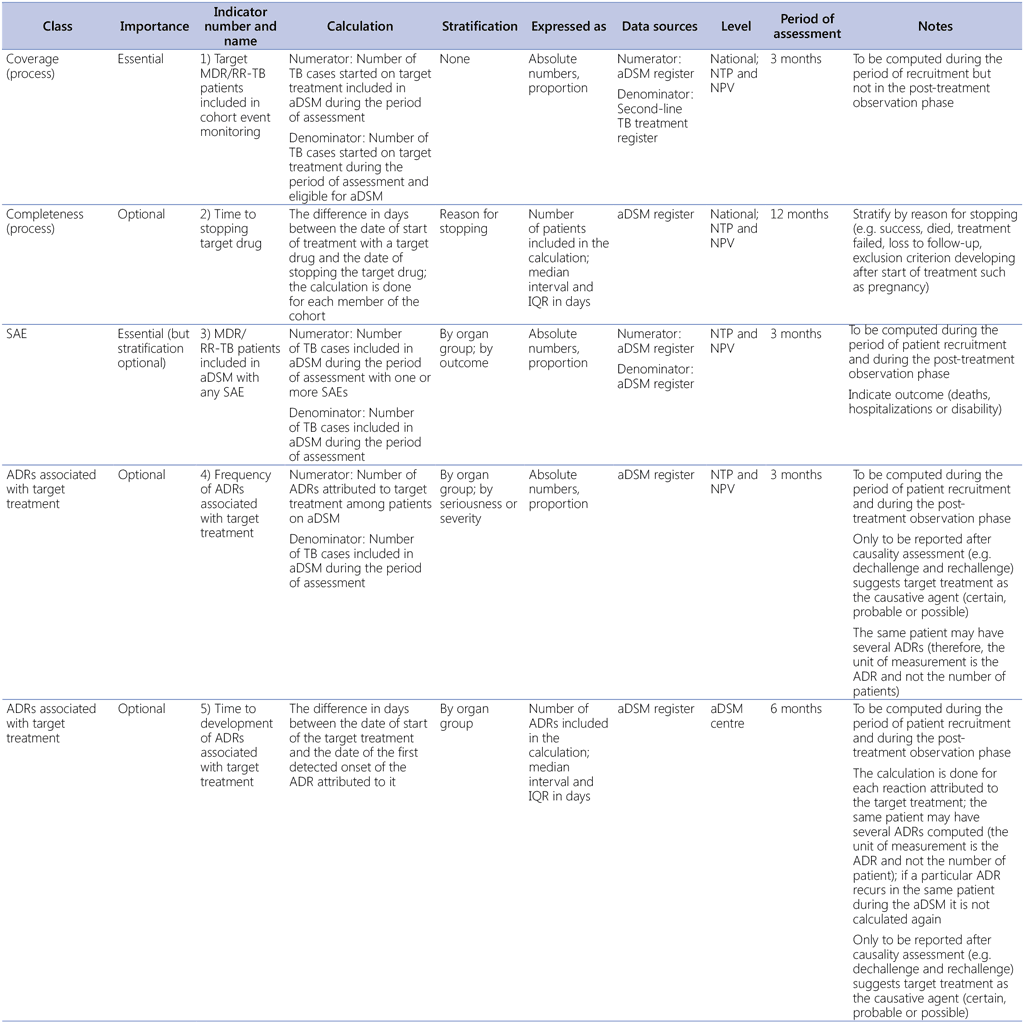
ADR: adverse drug reaction; aDSM: active TB drug-safety monitoring and management; IQR: interquartile range; MDR/RR-TB: multidrug-resistant or rifampicin-resistant TB; NPV: national pharmacovigilance centre; NTP: national TB programme; SAE: serious adverse event; TB: tuberculosis.
Adapted from WHO (2014) (7).
Table A3.2. Elements for a summary profile of drug safety or toxicity

aDSM: active TB drug-safety monitoring and management; MDR-TB: multidrug-resistant TB; TB: tuberculosis.
Adapted from the Draft framework for the harmonized and standardized summarization of both added benefit and risk associated with an intervention (7).
Appendix 1. List of contributors to the development of the aDSM framework

Appendix 2. Adverse events of clinical significance or special interest for aDSM
See Section A3.2 (Definitions used in aDSM) for the definitions of types of adverse events (AEs) mentioned in this appendix. AEs of clinical significance or special interest for active TB drug-safety monitoring and management (aDSM) are as follows:⁷
- All serious adverse events (SAEs).
- All AEs of special interest (suggested list):⁸
- peripheral neuropathy (paraesthesia);
- psychiatric disorders and central nervous system toxicity (e.g. depression, psychosis, suicidal intention and seizures);
- optic nerve disorder (optic neuritis) or retinopathy;
- ototoxicity (hearing impairment and hearing loss);
- myelosuppression (manifested as anaemia, thrombocytopenia, neutropenia or leukopenia);
- prolonged QT interval (Fridericia correction; see (7));
- lactic acidosis;
- hepatitis (defined as increases in alanine aminotransferase [ALT] or aspartate aminotransferase [AST] ≥5× the upper limit of normal [ULN], or increases in ALT or AST ≥3× ULN with clinical manifestations, or increases in ALT or AST ≥3× ULN with concomitant increase in bilirubin ≥1.5× ULN);
- hypothyroidism;
- hypokalaemia;
- pancreatitis;
- phospholipidosis; and
- acute kidney injury (acute renal failure).
- AEs leading to treatment discontinuation or change in drug dosage.
- AEs not listed above but judged as otherwise clinically significant by the clinician.
Appendix 3. Alert for serious adverse events to the TB programme (Example)
Confidential – to be sent even when there is suspicion of a serious adverse event (SAE).
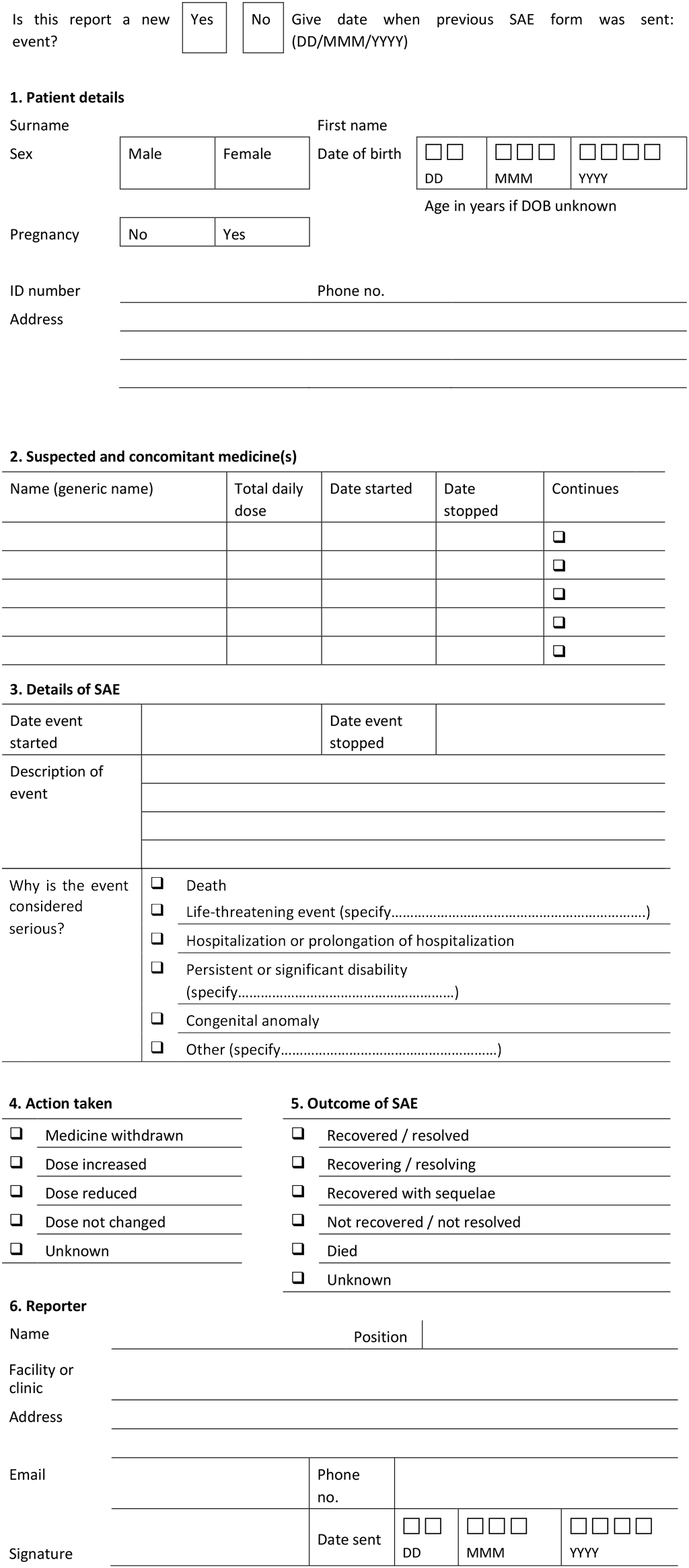
Confidential – to be sent even when there is suspicion of a serious adverse event (SAE).
Explanatory note – to be adapted according to the local situation:
- This form is intended for the core package of active tuberculosis (TB) drug-safety monitoring and management (aDSM). For more details, please refer to other documents on aDSM. The spontaneous reporting form in use by the national pharmacovigilance authorities may be adapted for the purposes of alerting the TB programme of SAEs and avoiding parallel reporting structures.
- The completed form can be sent electronically, via email or fax to <address> and the responsible authority alerted by phone.
- The report should be sent within <number> hours after it is detected, even when there is a suspicion of seriousness.
- The report should be sent even when not all details are available and the association with any particular medicine is uncertain. The essential details are the identifiers of the patient and the reporter; the name of the suspected medicine(s); and basic details on the SAE.
- If the report relates to a previously notified event, indicate this under Section 3 of the form; if more than one SAE occurs in the same individual, send separate forms for each event.
- All health care professionals are encouraged to report; patients and relatives may also report.
- Upon receipt of the information, the responsible authority will review the information and contact the reporter or the facility (or both) for more details. All information, including the identity of the patient and reporter, must be handled in strict confidence. Apart from action to protect public health, anonymized statistics from these reports will be used to improve drug safety.
- When reporting, use the format DD/MMM/YYYY to report dates. Under “Description of event” in Section 3 of the form, provide a single diagnosis and include anatomical location if applicable. If the diagnosis is unknown, describe the clinical picture.
References (Annex 3)
- Inter-regional workshop on pharmacovigilance for new drugs and novel regimens for the treatment of drug-resistant tuberculosis. Meeting Report. Hanoi, Viet Nam. 12–14 November 2014. Geneva: 2014 (https://www.who.int/publications/m/item/inter-regional-workshop-on-pharmacovigilance-for-new-drugs-andnovel-regimens-for-the-treatment-of-drug-resistant-tuberculosis, accessed 12 December 2022).
- Active tuberculosis drug-safety monitoring and management (aDSM): framework for implementation. Geneva: World Health Organization; 2015 (https://apps.who.int/iris/bitstream/handle/10665/204465/WHO_HTM_TB_2015.28_eng.pdf, accessed 1 June 2020).
- Policy implementation package for new TB drug introduction. Geneva: World Health Organization; 2014 (https://apps.who.int/iris/handle/10665/180199, accessed 12 December 2022).
- The safety of medicines in public health programmes: Pharmacovigilance an essential tool. Geneva: World Health Organization; 2006 (http://apps.who.int/iris/bitstream/handle/10665/43384/9241593911_eng.pdf, accessed 21 September 2022).
- Guideline on good pharmacovigilance practices (GVP). Annex 1 – Definitions (Rev 4). European Medicines Agency & Heads of Medicines Agencies; 2017 (https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-annex-i-definitions-rev-4_en.pdf, accessed 21 September 2022).
- Guideline for Industry – Clinical safety data management: definitions and standards for expedited reporting. FDA Center for Drug Evaluation and Research; 1995 (http://www.fda.gov/downloads/Drugs/Guidances/ucm073087.pdf, accessed 21 September 2022).
- Companion handbook to the WHO guidelines for the programmatic management of drug- resistant tuberculosis. Geneva: World Health Organization; 2014 (https://apps.who.int/iris/bitstream/handle/10665/130918/9789241548809_eng.pdf, accessed 11 October 2022).
- Pharmacovigilance guideline for endTB Projects outside Interventional Clinical Trial. version 0.7. UNITAID, Partners in Health, Médecins sans Frontières, Interactive Research & Development; 2015.
5 The definitions of some terms have been modified from those in general usage, to fit better in the context of national TB programmes (NTPs).
6 Countries and stakeholders may also monitor other AEs of special interest or clinical significance (see next section).
7 List adapted from Pharmacovigilance guideline for endTB projects outside interventional clinical trial, version 0.7 (8).
8 The list shown here is provisional; it may be modified according to the composition of the regimen or the patient cohort.
