
 Feedback
FeedbackRecommendation 1.1 The 6-month bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) regimen

Remarks
- Drug susceptibility testing (DST) for fluoroquinolones is strongly encouraged in people with MDR/RR-TB, and although it should not delay initiation of the BPaLM, results of the test should guide the decision on whether moxifloxacin can be retained or should be dropped from the regimen – in cases of documented resistance to fluoroquinolones, BPaL without moxifloxacin would be initiated or continued.
- This recommendation applies to the following:
- People with MDR/RR-TB or with MDR/RR-TB and resistance to fluoroquinolones (pre-XDR-TB).
- People with confirmed pulmonary TB and all forms of extrapulmonary TB except for TB involving the CNS, osteoarticular or disseminated forms of TB with multiorgan involvement.²¹
- Adults and adolescents aged 14 years and older.
- All people regardless of HIV status.
- Patients with less than 1-month previous exposure to bedaquiline, linezolid, pretomanid or delamanid. When exposure is greater than 1 month, these patients may still receive these regimens if resistance to the specific medicines with such exposure has been ruled out.
- This recommendation does not apply to pregnant and breastfeeding women owing to limited evidence on the safety of pretomanid.²²
- The recommended dose of linezolid is 600 mg once daily, both for the BPaLM and the BPaL regimen.²³
Rationale
The rationale for this recommendation is based on the evidence and considerations described in detail in the following two subsections. Briefly, data from an RCT (stage 2 of TB-PRACTECAL, corresponds to a Phase 3 trial) showed much improved treatment success rates with the BPaLM regimen (89%) of 6 months duration compared with the current SoC regimens (52%), as well as lower levels of treatment failure, death and loss to follow-up (LTFU). Data from two trials (TB-PRACTECAL and ZeNix) suggested fewer AEs with a linezolid dose of 600 mg while maintaining high efficacy. It was judged that implementing this regimen was probably feasible and acceptable, with cost–effectiveness and equity probably improved. The comparison of patient groups receiving this regimen with those receiving currently recommended regimens lasting 9 months or longer has favoured the 6-month BPaLM regimen, suggesting it to be the regimen of choice for eligible patient groups.
Summary of evidence
This section provides the PICO questions posed, the data and studies considered to answer the questions, the methods used for analysis and data synthesis, a summary of evidence on desirable and undesirable effects and certainty of evidence, and a summary of other evidence considered during the recommendation’s development. Additional detail on the evidence is available in the annexes containing the GRADE evidence summary tables and GRADE EtD tables (Annex 5).
PICO questions
The recommendation in this section is a result of assessments of the PICO questions listed below. Because of the different intervention and comparator groups used, PICOs 3, 5, and 6 have been split into several sub-PICO questions (details are given in the text and in Table 1.3).
PICO question 3–2022 (MDR/RR-TB, 2022): Should BPaL regimens with lower linezolid exposure (dose or duration) be used instead of the original BPaL regimen in patients who are eligible for BPaL regimen?
PICO question 4–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid, linezolid be used in patients with pulmonary pre-XDR-TB (MDR/RR-TB with fluoroquinolone resistance)?
PICO question 5–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid and linezolid be used in patients with pulmonary MDR/RR-TB and without fluoroquinolone resistance?
PICO question 6–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid and linezolid with or without addition of moxifloxacin (BPaLM) or clofazimine be used in patients with pulmonary MDR/RR-TB (with or without fluoroquinolone resistance)?
Data and studies considered
The review of this group of PICO questions during the GDG meeting convened by WHO in February– March 2022 was based on new evidence provided by MSF from the TB-PRACTECAL clinical trial and by the TB Alliance from the ZeNix trial. For several assessments under this PICO question, the data from the 2021 WHO individual patient dataset (IPD) were used. Patient populations included in two trials were recruited following strict inclusion and exclusion criteria; the populations had many similarities and few notable differences. The highlights of the criteria used by these trials are presented in Table 1.1. For a complete list of the exclusion criteria, see published trial protocols.²⁴
Table 1.1. High-level summary of main inclusion and exclusion criteria: TB-PRACTECAL and ZeNix trials
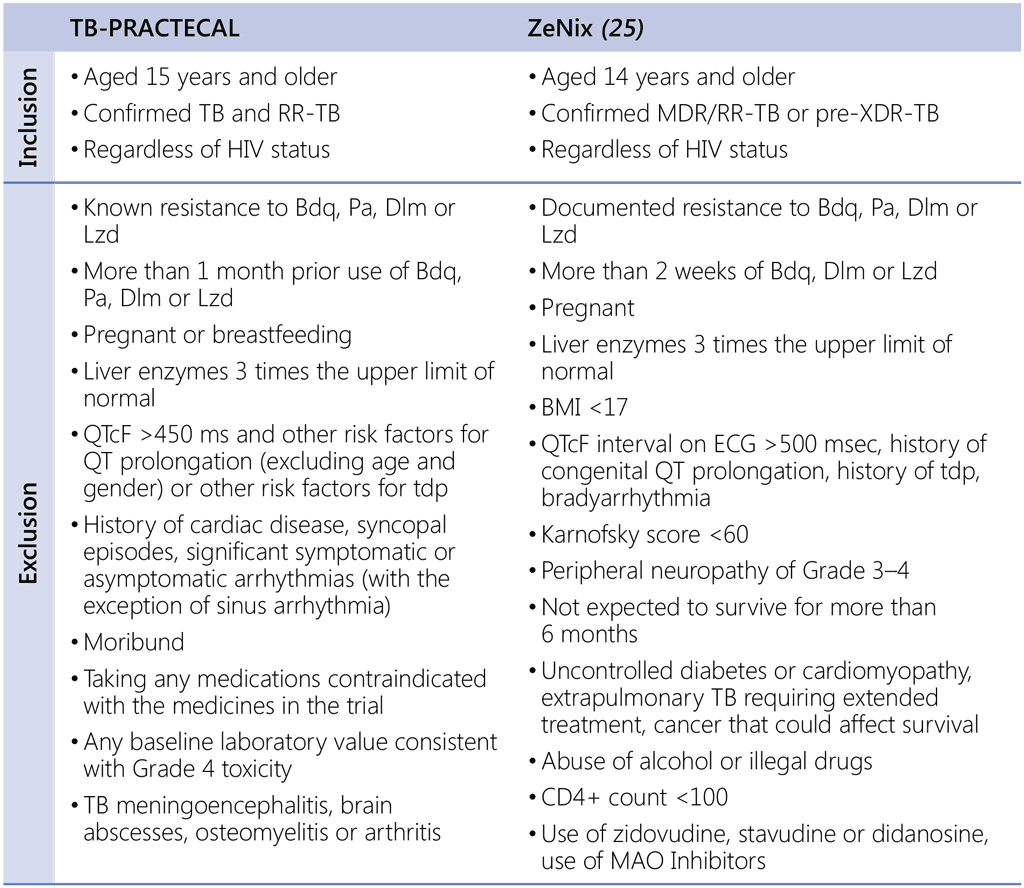
Bdq: bedaquiline; BMI: body mass index; Dlm: delamanid; ECG: electrocardiogram; HIV: human immunodeficiency virus; Lzd: linezolid; MAO: monoamine oxidase; MDR/RR-TB: multidrug-resistant or rifampicin-resistant TB; P: rifapentine; QTcF: corrected QT interval by Fredericia; RR-TB: rifampicin-resistant TB; TB: tuberculosis; tdp: torsades de pointes; pre-XDR-TB: pre-extensively drug-resistant TB.
TB-PRACTECAL
TB-PRACTECAL was a multicentre, open-label, multi-arm, randomized, controlled, multistage, Phase 2–3 trial evaluating short treatment regimens containing bedaquiline and pretomanid in combination with existing and repurposed anti-TB drugs (e.g. linezolid and clofazimine) for the treatment of microbiologically confirmed pulmonary MDR/RR-TB.²⁵
The study was divided into two stages, with a seamless transition between the stages, meaning that recruitment into an arm would only stop after a decision had been taken following stage 1 primary endpoint data analysis. In the first stage – equivalent to a Phase 2B trial of safety and preliminary efficacy – patients were randomly assigned one of four regimens, stratified by site. Investigational regimens included oral bedaquiline, pretomanid and linezolid. Two of the regimens also included moxifloxacin (arm 1) and clofazimine (arm 2). The main objective of Stage 1 was to select drug regimens for evaluation in stage 2, based on 8-week safety and efficacy endpoints. Investigational arms that did not meet predefined safety and efficacy criteria were not considered for further evaluation.
The second stage of the study was equivalent to a Phase 3 trial investigating the safety and efficacy of the most promising regimen. As intended in the study protocol, the regimen was evaluated for safety and efficacy in comparison with the SoC arm at 72 weeks after randomization. Stage 2 of the trial included an intervention arm of BPaLM compared with the locally approved SoC, consistent with WHO recommendations for the treatment of MDR/RR-TB or pre-XDR-TB at the time of trial conduct (including a 9–12-month injectable-containing regimen; 18–24-month WHO-recommended regimen [pre-2019]; 9–12-month all-oral regimen; and 18–20-month all-oral regimen). The TB-PRACTECAL trial stopped enrolling patients soon after its independent data safety and monitoring board indicated that the BPaLM regimen is superior to the SoC, because it was considered that more data were extremely unlikely to change the results of the trial. This trial was not designed to compare the investigational regimens against each other.
Eligible patients were aged 15 years and older, and had bacteriologically (molecular or phenotypic) confirmed TB and resistance to at least rifampicin by a molecular or phenotypic drug susceptibility test. The primary efficacy outcome was the composite endpoint of unfavourable outcomes (failure, death, treatment discontinuation, recurrence or LTFU) at 72 weeks after randomization. Relevant secondary efficacy outcomes included culture conversion at 12 and 24 weeks, unfavourable outcomes at 24 weeks after randomization, unfavourable outcomes at 108 weeks after randomization, median time to culture conversion and recurrence by week 48 in the investigational arms. Participants were randomized in a 1:1:1:1 ratio into either the SoC or one of the following three intervention arms:
- Arm 1: 24 weeks of B-Pa-Lzd-Mfx (BPaLM);
- Arm 2: 24 weeks of B-Pa-Lzd-Cfz (BPaLC); and
- Arm 3: 24 weeks of B-Pa-Lzd (BPaL).
In all intervention arms, linezolid was given at 600 mg daily for 16 weeks then 300 mg daily for the remaining 8 weeks (or earlier when moderately tolerated). Bedaquiline was given at 400 mg once daily for 2 weeks followed by 200 mg three times per week for 22 weeks. Safety monitoring for most participants included multiple electrocardiograms (ECGs) at baseline, then weekly until week 8, every 4 weeks up to week 24 and then every 8 weeks thereafter. Microbiological monitoring included smear microscopy and culture at baseline and day 7, then every 4 weeks up until week 24 and every 8 weeks thereafter.
ZeNix
ZeNix was a Phase 3 partially blinded, randomized trial assessing the safety and efficacy of various doses and treatment durations of linezolid plus bedaquiline and pretomanid in individuals with pulmonary MDR/RR-TB and additional resistance to fluoroquinolones (with or without resistance to injectable agents) or those with treatment intolerant or nonresponsive MDR/RR-TB. Eligible patients were aged 14 years and older, weighed at least 35 kg, had a documented HIV result and had bacteriologically confirmed sputum culture positive XDR-TB (pre-2021 definition) or bacteriologically confirmed MDR/RR-TB, but were treatment intolerant or nonresponsive to previous MDR/RR-TB treatment. The primary study outcome was the incidence of bacteriological failure or relapse or clinical failure through follow-up until 26 weeks after the end of treatment. The secondary outcomes included incidence of bacteriological failure or relapse or clinical failure through follow-up until 78 weeks after the end of treatment. Participants received 26 weeks of treatment with BPaL. Each of the four arms varied the dose and duration of linezolid: 1200 mg 26 weeks, 1200 mg 9 weeks, 600 mg 26 weeks or 600 mg 9 weeks. Bedaquiline was given at 200 mg once daily for 8 weeks then 100 mg once daily for 18 weeks. This off-label dosing schedule is supported by pharmacokinetic simulations for an alternative bedaquiline dosing schedule that provides comparable exposures and was developed to support adherence and facilitate treatment administration (all medicines daily throughout the regimen) (31). Safety monitoring included scheduled testing and assessments of laboratory parameters, ECG, vital signs and other physical examinations (32). Microbiological monitoring included smear microscopy, molecular testing and liquid culture from sputum at baseline and liquid culture at all patient visits thereafter (32).
Table 1.2. Dosing, treatment administration and toxicity-related treatment modification tolerances


2021 WHO IPD
In 2021, WHO issued a public call for data to serve as a comparator group (SoC) against which 6–9-month regimens could be compared. These cohorts received treatment conforming to the WHO DR-TB guidelines of 2020 with bedaquiline and linezolid for a duration ranging from 6 to 24 months. Patients receiving injectable antibiotics were excluded.
Included datasets comprised individuals using one of the following regimens:
- 6–12-month all-oral regimens using at least bedaquiline and linezolid; or
- 9–12-month WHO (2019) all-oral bedaquiline-containing regimen in the combination, such as 4–6 Bdq(6m)-Lfx/Mfx-Cfz-Z-E-Hh-Eto/5m Lfx/Mfx-Cfz-Z-E; or
- ≥18-month WHO (2018) all-oral treatment regimen containing at least bedaquiline and linezolid.
The individual datasets that are included in this cohort are described in detail in the statistical analysis plan (Annex 6). To be eligible for inclusion in a short comparator regimen (target 9–12 months at treatment commencement), patients must have fulfilled each of the following:
- had a treatment duration not exceeding 12 months;
- received six or more drugs during treatment, including bedaquiline; and
- if given an outcome of cure or completed, had a treatment duration of 8.5 months or more.
To be eligible for inclusion in a longer comparator regimen (target 18–24 months), patients must have fulfilled each of the following:
- be classified in the dataset as having received a longer regimen (if stated);
- had a treatment duration not longer than 24 months;
- received four or more drugs (regardless of drug susceptibility; i.e. regardless of whether they were likely to be effective), including bedaquiline; and
- if given an outcome of cure or complete, had a treatment duration of 17.5 months or more.
Methods used for analysis and data synthesis
Descriptive analyses of the baseline characteristics of participants in all included studies were performed; characteristics included demographics, diagnostic test results, treatment regimens and treatment outcomes.
Comparative analyses were performed within individual studies and between multiple studies:
- Within study comparisons – for studies in which both a short-course (6 months in duration) regimen and a relevant comparator are used, pairwise comparisons were conducted between each of the short-course regimens and the comparator. For included RCTs (e.g. the TB-PRACTECAL trial and NExT trial), the primary outcome of the prespecified analysis was also calculated and reported.
- Pairwise comparisons between studies – comparisons addressing each PICO question were conducted by comparing outcomes among cohorts in which participants received either the intervention or the control regimen relevant to that question.
Statistical models
For comparisons between dataset or cohorts, outcomes were presented as unadjusted and adjusted risk ratios (RR). Adjusted risk ratios (aRR) were calculated using a log-binomial generalized linear regression (binomial error distribution with log link function). Pre-specified potential confounders were adjusted for using inverse probability propensity score weighting. No convergence issues arose with the log-binomial model. When outcome rates were close to the boundary, aRR were not calculated, and unadjusted RR were presented. For outcomes where the number of outcome events was zero, an unadjusted risk difference (RD) was calculated. For unadjusted RDs or RRs, 95% confidence intervals (CIs) were calculated using the score method. Covariate selection for calculation of propensity scores was based on data availability and clinical knowledge. The covariates considered for inclusion in the propensity scores analysis included age, gender, baseline smear result, HIV status (including ART status), prior treatment history (including whether previous infection was drug resistant), body mass index (BMI), smoking status, diabetes diagnosis, cavitation at baseline, presence of bilateral disease and fluroquinolone resistance. For the calculation of aRRs, multiple imputation by chain equations using the “within” propensity score approach was used to account for missing data in potential confounders when the proportion of missing values for a confounder was less than 45%.
Timing of follow-up for comparisons between regimens
The analyses undertaken for this evidence review combined results from cohorts with differing follow-up times after initiation of treatment. There were differences in the follow-up time between cohorts (from 5.5 months to 24 months) and within single cohorts (e.g. the WHO IPD 2021 dataset combined multiple cohorts with variable follow-up times). Follow-up time was separated into the time between commencement of treatment and treatment completion, and the period from treatment completion until the end of follow-up. For shorter regimens, post-treatment follow-up was particularly important because higher relapse rates may be a consequence of shorter treatments that do not completely remove M. tuberculosis. Where possible, it was important for follow-up time between two groups in a comparison to be equivalent, so that participants had an equivalent likelihood of death or relapse. In these analyses, the follow-up time was measured from the start date of treatment rather than after the date of treatment completion, to minimize the effect of differences in total follow-up time.
The principles for accounting for time periods of follow-up were as follows:
- Where possible, follow up participants in the intervention and control groups for the same total time, so that the likelihood of unsuccessful outcomes (e.g. death) is the same in both groups.
- Limit follow-up to 24 months after treatment initiation for all cohorts. There were no analyses in which both intervention and comparator cohorts had more than 24 months of follow-up available. The evidence accumulated from TB treatment trials demonstrates that a high proportion of recurrences are likely to occur within 12 or even 6 months of stopping treatment (33).
- Select a primary analysis that optimizes the number of participants included in both groups. For shorter (6–9-month regimens), follow-up time in the comparison was included to allow for relapse to be captured.
Additional sensitivity analyses were performed, where possible, evaluating the effect of follow-up time upon treatment outcomes.
Summary of evidence on desirable and undesirable effects and certainty of evidence
The evidence on the novel regimens to inform PICO questions was derived from two trials. It included information on a total of 419 of 423 participants who were enrolled in four arms of the TB-PRACTECAL and on 172 of 181 participants who were enrolled in four arms of the ZeNix trial²⁶
Data from patients in relevant arms of these trials were used in each of the comparisons that led to the conclusions and final recommendation on the use of the BPaLM/BPaL regimen. Even though the TB-PRACTECAL trial was not designed to compare the investigational regimens against each other and with the SoC, the comparisons of the different arms of the trial to the BPaLM arm (sub-PICOs 6.2 to 6.6) were performed to aid the panel in making final decisions.
Sub-PICO 3.2
The BPaL 1200–9 arm of the ZeNix trial (where linezolid 1200 mg daily was used for 9 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 1200–9 (n=43) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- lower levels of treatment success (93% vs 98%); that is, a 5% relative reduction (RR=0.95, 95% CI: 0.87 to 1.05);
- higher levels of failure and recurrence (4.7% vs 2.3%); that is, a twofold relative increase (RR=2.1, 95% CI: 0.19 to 22);
- higher levels of deaths (2.3% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.06 to 0.12);
- the same levels of LTFU (0% vs 0%); that is, a 0% absolute difference (RD=0.00, 95% CI: –0.08 to 0.08);
- lower levels of AEs (16% vs 18%); that is, a 10% relative reduction (RR=0.90, 95% CI: 0.36 to 2.3); and
- the same levels of amplification of drug resistance (0% vs 0%); that is, a 0% absolute difference (RD=0.00, 95% CI: –0.08 to 0.08).
The GDG judged the benefits of BPaL with linezolid 1200–9 to be small and the undesirable effects to be moderate compared with BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 1200–26.
Conclusion
The use of the 26 weeks of 1200 mg linezolid is suggested over 9 weeks of 1200 mg linezolid as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
Sub-PICO 3.3
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–26 (n=43) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- higher levels of treatment success (100% vs 98%); that is, a 2% relative increase (RR=1.02, 95% CI: 0.98 to 1.07);
- lower levels of failure and recurrence (0% vs 2.3%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.12 to 0.06);
- lower levels of Grade 3–5 AEs (14% vs 18.6%); that is, a 23% relative reduction (RR=0.77, 95% CI: 0.29 to 2.03); and
- the same levels of deaths (0% vs 0%), LTFU (0% vs 0%) or amplified resistance (0% vs 0%).
The GDG judged the benefits of BPaL with linezolid 600–26 to be moderate and the undesirable effects to be trivial compared with BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 26 weeks of 600 mg linezolid over 26 weeks of 1200 mg linezolid is suggested as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
Sub-PICO 3.4
The BPaL 600–9 arm of the ZeNix trial (where linezolid 600 mg daily was used for 9 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–9 (n=42) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- lower levels of treatment success (93% vs 98%); that is, a 5% relative reduction (RR=0.95, 95% CI: 0.86 to 1.05);
- higher levels of failure and recurrence (4.8% vs 2.3%); that is, a twofold increase (RR=2.10, 95% CI: 0.20 to 22.26);
- higher levels of LTFU (2.4% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.06 to 0.12);
- lower levels of Grade 3–5 AEs (14.3% vs 18.2%); that is, a 21% relative reduction (RR=0.79, 95% CI: 0.30 to 2.07); and
- the same levels of deaths (0% vs 0%) or amplified resistance (0% vs 0%).
The GDG judged the benefits of BPaL with linezolid 600–9 to be small and the undesirable effects to be moderate compared with the BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 1200–26.
Conclusion
The use of the 26 weeks of 1200 mg over 9 weeks of 600 mg linezolid is suggested as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
PICO 3 – Intermediate summary conclusion
The assessment of PICO 3 allowed for the decision on the optimal dosing and duration of linezolid within the BPaLM/BPaL regimen, and narrowed down the subsequent comparisons to the intervention regimen with this particular dose and duration of linezolid – BPaL (600 mg – 26 weeks).
Sub-PICO 4.1
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients with fluoroquinolone resistance from the 2021 IPD who were receiving longer regimens for treatment of MDR/RR-TB, designed in line with 2020 WHO guidelines. Primary analysis was undertaken at 18 months post treatment initiation.
Participants with pulmonary pre-XDR-TB (MDR/RR-TB with fluoroquinolone resistance) receiving BPaL 600–26 (n=33) compared with participants receiving longer regimens for MDR/RR-TB (n=839) experienced:
- higher levels of treatment success (100% vs 75%); that is, a 34% relative increase (RR=1.34, 95% CI: 1.20 to 1.40);
- lower levels of failure and recurrence (0% vs 6.6%); that is, a 7% absolute reduction (RD= –0.07, 95% CI: –0.08 to –0.04);
- lower levels of deaths (0% vs 9.9%); that is, a 10% absolute reduction (RD= –0.10, 95% CI: –0.12 to –0.01);
- lower levels of LTFU (0% vs 9.1%); that is, a 9% absolute reduction (RD= –0.09, 95% CI: –0.11 to –0.01); higher levels of AEs (15% vs 4.4%); that is, a 3.4-fold increase (RR=3.44, 95% CI: 1.44 to 8.17); and
- lower levels of amplification of drug resistance (0% vs 7.4%); that is, a 7% absolute reduction (RD= –0.07, 95% CI: –0.09 to –0.03).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with longer regimens recommended by WHO. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than a longer (18-month) regimen is suggested in patients with MDR/RR-TB and resistance to fluoroquinolones (pre-XDR-TB), who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 4 – Intermediate conclusion
The assessment of PICO 4 resulted in the conditional recommendation for use of the BPaL (600 mg – 26 weeks) regimen over the currently recommended longer regimens in patients with MDR/RR-TB and additional fluoroquinolone resistance (pre-XDR-TB).
Sub-PICO 5.1
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance treated in South Africa with the WHO-recommended 9-month regimen with ethionamide for 4 months. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaL 600–26 regimen (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving the 9-month regimen with ethionamide (n=785) experienced:
- higher levels of treatment success (100% vs 69%); that is, a 45% relative increase (RR=1.45, 95% CI: 1.32 to 1.53);
- lower levels of failure and recurrence (0% vs 1.3%); that is, a 1% absolute reduction (RD=–0.01, 95% CI: –0.02 to 0.07);
- lower levels of deaths (0% vs 19%); that is, a 19% absolute reduction (RD=–0.19, 95% CI: –0.22 to –0.1);
- lower levels of LTFU (0% vs 11%); that is, an 11% absolute reduction (RD= –0.11, 95% CI: –0.14 to –0.03); and
- the same levels of amplified resistance (0% vs 0%); that is, a 0% absolute difference (RD= 0.00, 95% CI: –0.01 to 0.08).
Grade 3–5 AEs were noted in 14% of participants receiving the BPaL 600–26 but no comparison could be done because no data were available for participants receiving the 9-month regimen with ethionamide.
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with the WHO-recommended 9-month regimen with ethionamide. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than the 9-month regimen (with ethionamide) is suggested in patients with MDR/RR-TB without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 5.2
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance from the 2021 IPD, treated with longer regimens for MDR/RR-TB, designed in line with the 2020 WHO guidelines. Primary analysis was undertaken at 18 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL 600–26 regimen (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving longer regimens recommended by WHO (n=850) experienced:
- higher levels of treatment success (98% vs 74%); that is, a 32% relative increase (RR=1.32, 95% CI: 1.19 to 1.39);
- lower levels of failure and recurrence (2.3% vs 3.3%); that is, a 29% relative reduction (RR=0.71, 95% CI: 0.12 to 3.8);
- lower levels of deaths (0% vs 11%); that is, an 11% absolute reduction (RD= –0.11, 95% CI: –0.13 to –0.03);
- lower levels of LTFU (0% vs 12%); that is, a 12% absolute reduction (RD= –0.12, 95% CI: –0.14 to –0.04);
- higher levels of Grade 3–5 AEs (14% vs 5%); that is, a fourfold relative increase (aRR=3.99, 95% CI: 1.67 to 9.57); and
- lower levels of amplified resistance (0% vs 2.4%); that is, a 2% absolute decrease (RD= –0.02, 95% CI: –0.04 to 0.06).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with longer regimens recommended by WHO. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than longer (18-month) regimens is suggested in patients with MDR/RR-TB and without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 5.3
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance treated in South Africa with a 9-month regimen with linezolid for 2 months. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–26 (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving a 9-month regimen with linezolid (n=4216) experienced:
- higher levels of treatment success (100% vs 66%); that is, a 52% relative increase (RR=1.52, 95% CI: 1.38 to 1.55);
- lower levels of failure and recurrence (0% vs 1.2%); that is, a 1% absolute reduction (RD= –0.01, 95% CI: –0.02 to 0.07);
- lower levels of deaths (0% vs 18%); that is, an 18% absolute reduction (RD= –0.18, 95% CI: –0.19 to –0.1);
- lower levels of LTFU (0% vs 15%); that is, a 15% absolute reduction (RD= –0.15, 95% CI: –0.16 to –0.07);
- higher levels of Grade 3–5 AEs (14% vs 4.9%); that is, a threefold increase (aRR=2.92, 95% CI: 1.38 to 6.18); and
- lower levels of amplified resistance (0% vs 0.6%); that is, a 1% absolute reduction (RD= –0.01, 95% CI: –0.01 to 0.08).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with receiving a 9-month regimen with linezolid. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than the 9-month regimen (with linezolid) is suggested in patients with MDR/RR-TB without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 5 – Intermediate summary conclusion
The three assessments performed under PICO 5 resulted in the conditional recommendations for the BPaL (600 mg – 26 weeks) regimen over the currently recommended 9-month regimen with ethionamide (sub-PICO 5.1), over longer (18-month) regimens (sub-PICO 5.2) and over the new 9-month regimen where ethionamide is replaced with 2 months of linezolid (subPICO 5.3) in patients with pulmonary MDR/RR-TB without fluoroquinolone resistance.
Sub-PICO 6.1
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the comparator arm of the TB-PRACTECAL trial, which comprised MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens recommended by WHO at the time the trial was conducted (including a 9–12-month injectable-containing regimen, an 18–24-month WHO-recommended regimen [pre-2019], a 9–12-month all-oral regimen and an 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher levels of treatment success (89% vs 52%); that is, a 73% relative increase (aRR=1.73, 95% CI: 1.31 to 2.27);
- lower levels of failure and recurrence (8% vs 26%) that is 74% relative reduction (aRR=0.26, 95% CI: 0.10 to 0.71);
- lower levels of deaths (0% vs 3.0%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.10 to 0.03);
- lower levels of LTFU (3.2% vs 20%); that is, a 84% relative reduction (RR=0.16, 95% CI: 0.04 to 0.61);
- lower levels of Grade 3–5 AEs (21% vs 51%); that is, a 59% relative reduction (aRR=0.41, 95% CI: 0.26 to 0.63); and
- lower levels of amplified resistance (0% vs 1.9%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.07 to 0.02).
The GDG judged the benefits of BPaLM to be large and the undesirable effects to be trivial compared with WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours the BPaLM regimen.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than a 9-month or longer (18-month) regimen is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.2
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaL arm of the TB-PRACTECAL trial, which comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving BPaL in the TB-PRACTECAL trial (n=60) experienced:
- higher levels of treatment success (89% vs 77%); that is, a 15% relative increase (aRR=1.15, 95% CI: 0.95 to 1.38);
- lower levels of failure and recurrence (8.1% vs 13%); that is, a 47% relative reduction (aRR= 0.53, 95% CI: 0.17 to 1.63);
- lower levels of LTFU (3.2% vs 10%); that is, a 68% relative reduction (aRR=0.32, 95% CI: 0.08 to 1.34);
- no difference in deaths (0% vs 0%); that is, a 0% absolute difference (RD= 0, 95% CI: –0.06 to 0.06);
- higher levels of Grade 3–5 AEs (21% vs 20%); that is, a 7% relative increase (aRR=1.07, 95% CI: 0.62 to 1.88); and
- lower levels of amplified resistance (0% vs 2.9%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.08 to 0.01).
The GDG judged the benefits of BPaLM to be moderate and the undesirable effects to be small compared with BPaL. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLM.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than BPaL is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.3
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaLC arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving the BPaLC regimen (n=64) in the TB-PRACTECAL trial experienced:
- higher levels of treatment success (89% vs 81%); that is, an 11% relative increase (aRR 1.11, 95% CI: 0.94 to 1.31);
- lower levels of failure and recurrence (8.1% vs 9.4%); that is, a 30% relative reduction (aRR= 0.70, 95% CI: 0.2 to 2.29);
- lower levels of deaths (0% vs 1.6%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.08 to 0.04);
- lower levels of LTFU (3.2% vs 7.8%); that is, a 59% relative reduction (RR=0.41, 95% CI: 0.09 to 1.77);
- lower levels of Grade 3–5 AEs (21% vs 34%); that is, a 39% relative reduction (aRR=0.61, 95% CI: 0.37 to 1.00); and
- lower levels of amplified resistance (0% vs 1.9%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.07 to 0.02).
The GDG judged the benefits of BPaLM to be moderate and the undesirable effects to be trivial compared with BPaLC. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLM.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than BPaLC is suggested in patients with MDR/RR-TB with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.4
The BPaLC regimen arm of the TB-PRACTECAL trial with population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared to the comparator arm of the TB-PRACTECAL trial comprised of MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens recommended by WHO at the time of trial conduct (including a 9–12-month injectable-containing regimen; 18–24-month WHO-recommended regimen [pre-2019]; 9–12-month all-oral regimen; and 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaLC (n=64) compared to participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher treatment success (81% vs 52%); that is, a 55% relative increase (aRR=1.55, 95% CI: 1.15 to 2.11);
- lower levels of failure and recurrence (9.4% vs 26%); that is, a 66% relative reduction (aRR=0.34, 95% CI: 0.14 to 0.87);
- lower levels of deaths (1.6% vs 3.0%); that is, a 48% relative reduction (RR=0.52, 95% CI: 0.07 to 3.85);
- lower levels of LTFU (7.8% vs 20%); that is, a 57% relative reduction (aRR=0.43, 95% CI: 0.15 to 1.23);
- lower levels of grade 3 to 5 AEs (34% vs 51%); that is, a 33% relative reduction (aRR=0.67, 95% CI: 0.46 to 0.97); and
- higher levels of amplified resistance (1.9% vs 1.9%); that is, a 4% relative increase (RR=1.04, 95% CI: 0.19 to 5.80).
The GDG judged the benefits of BPaLC to be large and the undesirable effects to be trivial compared to WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLC.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and clofazimine (BPaLC) rather than a 9-month or longer (18-month) regimen is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month (overruled by conclusions of sub-PICO 6.5 and sub-PICO 6.6).
Sub-PICO 6.5
The BPaLC regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaL arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with pulmonary MDR/RR-TB or pre-XDR-TB receiving BPaLC (n=64) compared with participants receiving BPaL 600–300 (n=60) experienced:
- higher levels of treatment success (81% vs 77%); that is, a 4% relative increase (aRR=1.04, 95% CI: 0.84 to 1.30);
- lower levels of failure and recurrence (9.4% vs 13%); that is, a 14% relative reduction (aRR=0.86, 95% CI: 0.28 to 2.69);
- higher levels of deaths (1.6% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.05 to 0.08);
- lower levels of LTFU (7.8% vs 10%); that is, a 28% relative reduction (aRR=0.72, 95% CI: 0.21 to 2.47);
- higher levels of AEs (34% vs 20%); that is, a 64% relative increase (aRR=1.64, 95% CI: 0.97 to 2.79); and
- lower levels of amplification of drug resistance (1.9% vs 2.9%); that is, a 35% relative reduction (RR=0.65, 95% CI: 0.13 to 3.21).
The GDG judged both the desirable and the undesirable effects of BPaLC to be small compared with BPaL. The certainty of evidence was judged to be very low. The balance of health effects did not favour either the intervention or the comparator; however, taking into consideration the higher cost of the regimen, increased pill burden, reduced acceptability due to skin discolouration and other potential adverse effects related to clofazimine without noticeable net benefit in terms of health effects, the panel judged against the intervention.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than BPaLC is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.6
The BPaL arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the comparator arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens (including a 9–12-month injectable-containing regimen, an 18–24-month WHO regimen [pre-2019], a 9–12-month all-oral regimen and an 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL (n=60) compared with participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher levels of treatment success (77% vs 52%); that is, a 47% relative increase (aRR=1.47, 95% CI: 1.09 to 1.99);
- lower levels of failure and recurrence (13% vs 26%); that is, a 48% relative reduction (aRR=0.52, 95% CI: 0.22 to 1.18);
- lower levels of deaths (0% vs 3.0%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.10 to 0.03);
- lower levels of LTFU (10% vs 20%); that is, a 40% relative reduction (aRR=0.60, 95% CI: 0.24 to 1.56);
- lower levels of AEs (20% vs 51%); that is, a 62% relative reduction (RR=0.38, 95% CI: 0.24 to 0.60); and
- higher levels of amplification of drug resistance (2.9% vs 1.9%); that is, a 59% relative increase (RR=1.59, 95% CI: 0.32 to 7.84).
The GDG judged the benefits of BPaL to be large and the undesirable effects to be trivial compared with WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours the BPaL regimen.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than a 9-month or longer (18-month) regimen is suggested in patients with MDR/RR-TB with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 6 – Intermediate summary conclusion
The main assessment that defined the overall decision was that of sub-PICO 6.1, which resulted in the conditional recommendation for use of the BPaLM regimen over the internal mix of SoC regimens conforming to the WHO recommendations on 9-month or longer regimens. The assessments of the investigational regimens against each other and with the SoC in sub-PICOs 6.2–6.6 helped the panel in making final decisions.
Summary of other evidence
Additional data reviewed by the GDG relevant to these PICO questions were a cost–effectiveness analysis, a study on the acceptability and likelihood of implementation of the BPaL regimen, modelled pharmacokinetic data based on the development of a pharmacokinetic toxicodynamic model, and a summary of data on potential reproductive toxicity of pretomanid. No additional research data were available during review of sub-PICO questions 3.2–3.5.
Pharmacokinetic data
Early data from the pharmacokinetics study embedded in the TB-PRACTECAL were presented to the GDG panel in one of the preparatory webinars. The final results of this sub-study were not available at the time of the assessment and could not be fully considered.
The pharmacokinetics of linezolid are highly variable, with efficacy and toxicity dependent on factors such as pathogen susceptibility, drug exposure and the combination of companion drugs. The toxicity of linezolid, especially when used at higher doses and longer durations, is a known phenomenon and various strategies have been suggested to reduce it. However, except for the data available from the ZeNix and TB-PRACTECAL trials, no other strategies have been tested in a trial environment.²⁷
Data on reproductive toxicity of pretomanid
New data on the safety of pretomanid based on hormone evaluations in four clinical trials and a paternity survey were assessed; these data have largely alleviated previous concerns about reproductive toxicities observed in animal studies,²⁸ suggesting that adverse effects on human male fertility are unlikely. A study assessing semen in men undergoing treatment that includes pretomanid is in progress and will address any remaining concerns. Below is a summary of preclinical and clinical data relevant to testicular toxicity of pretomanid:
- rodent toxicology studies – evidence of direct testicular toxicity;
- monkey toxicology studies – no evidence for direct testicular toxicity; abnormal sperm findings considered to be secondary to declining physical condition;
- hormone data from clinical studies – no changes in follicle stimulating hormone (FSH), luteinizing hormone (LH) and inhibin B, consistent with testicular toxicity;
- paternity survey – 44 children fathered by 38 men (12%) who participated in pretomanid studies of 4–6 months treatment duration; and
- semen study – ongoing study evaluating semen in men undergoing pretomanid treatment.
Resources required and cost–effectiveness
Estimated regimen costs (in adults) at GDF prices²⁹ are about US$ 688 for BPaL (600–26), US$ 716 for BPaLM (600–26), an average of US$ 771 for longer regimens (depends on length and composition) and US$ 535–557 for 9-month regimens. Data from three studies were available on more detailed analyses of resources required and cost–effectiveness; two of these studies compared the BPaL regimen with longer (18-month) regimens (34, 35) and one compared the BPaL, BPaLM and BPaLC regimens with longer (18-month) regimens and with the 9-month regimen with ethionamide (36). The applicability of the results from these studies varied by PICO and sub-PICO question, and the panel noted associated caveats when discussing these results (details available in the GRADE EtD tables in Annex 5). Overall, based on these three publications, estimates for comparative total cost (drugs and delivery) within country appear to be between 1.4-fold and 6-fold higher (longer regimens) or 1–18% higher (9-month regimens) than for BPaLM/BPaL. Thus, the panel judged that implementation of BPaLM/BPaL would probably to lead to large savings when replacing the longer (18-month) regimens and moderate savings when replacing the 9-month regimens.
The cost–effectiveness study (36) found that, in most settings, BPaLM/BPaL is cost saving, mainly because of reduced time in care and therefore reductions in numbers of outpatient visits, inpatient bed-days and laboratory tests. The panel judged that cost–effectiveness probably favours BPaLM/BPaL.
Equity, acceptability and feasibility
The panel considered the treatment duration and the ability to decentralize treatment (to enable access for remote, underserved settings and disadvantaged populations) to affect equity. Despite not being able to identify relevant research evidence, the panel used their collective experience to judge that there would probably be advantages associated with the use of the BPaLM/BPaL regimen owing to its reduced complexity and shorter duration. Therefore, the panel judged that use of the BPaLM/BPaL regimen would probably increase equity.
A study on the acceptability and feasibility of the BPaL regimen from the provider perspective (37) was considered to be relevant evidence for the assessment of BPaL and indirectly for the assessment of BPaLM. This was a mixed-methods study among a cross section of health care workers, and programmatic and laboratory stakeholders that was carried out between May 2018 and May 2019 in Indonesia, Kyrgyzstan and Nigeria. The results from this study suggested that acceptability and feasibility overall were high. BPaL was rated as acceptable by more than 80% of participants across domains and stakeholders and 88% of interviewed stakeholders stated that they would probably implement BPaL once it became available. Stakeholders appreciated that BPaL would reduce the workload and financial burden on the health care system; expressed concerns about BPaL safety (monitoring), long-term efficacy and national regulatory requirements; and stressed the importance of addressing current health systems constraints, especially in treatment and safety monitoring systems. Results from a second qualitative study (38) with a focus on the patient perspective were presented to the panel; this study suggested that patients would welcome the positive impact of shorter treatment on employment status.³⁰
The panel noted these study results and, as part of their deliberations, they considered patients and health care providers as key stakeholders. The panel considered the following aspects to be critical with regard to the acceptability of BPaLM/BPaL: regimen duration and drug-safety monitoring needs (relating both to the necessary travel, loss of income and general disruption of the life of patients, and to workload for the health care system), and the need for DST. The panel judged that the BPaLM/BPaL regimen would probably be acceptable. Regarding feasibility, the panel noted the limited availability of pure substances of drugs in the BPaLM/BPaL regimen for use in DST as a potential barrier to implementation; they also noted that data on the critical concentration of pretomanid for use in DST are limited. However, given the reduced duration, complexity and associated workload of BPaLM/BPaL, the panel judged that implementation of BPaLM/BPaL is probably feasible.
Evidence to recommendations: considerations
Based on the decisions taken during the review and the combination of assessments described above, the recommendation is to use the BPaLM regimen as the first choice in the defined patient group with MDR/RR-TB, with the regimen to be used under routine programmatic conditions. Patients with MDR/RR-TB who cannot use the BPaLM regimen, either due to ineligibility or unavailability of its components, can instead be treated with 6-month BDLLfxC (see Recommendation 1.2) or one of the 9-month regimens (see Treatment of drug-resistant TB using 9-month regimens). The use of the longer regimens is reserved (see Treatment of drug-resistant TB using longer regimens) for individuals with MDR/RR-TB and fluoroquinolone resistance with further resistance or intolerance to bedaquiline, linezolid (XDR-TB) or pretomanid, and those with complicated extrapulmonary TB who would then receive a longer regimen designed with remaining effective medicines from Groups A, B and C, according to their drug susceptibility profile and other parameters.
Table 1.3 lists the comparisons and decisions on each of the sub-PICO-questions that were eventually used by the GDG to conclude with this summary recommendation. Throughout the discussion, the GDG panel focused on direct (within trial) comparisons among the TB-PRACTECAL trial arms, to ensure consistency and because it was felt that results based on random allocation to interventions were far more reliable than indirect, nonrandomized comparisons. Whereas the certainty of evidence of these (TB-PRACTECAL-internal) comparisons was still judged to be very low, the panel deemed it to be higher than that of other (indirect or between-trial or cohort) comparisons.
Although assessments of PICO questions 3, 4, 5 and 6 have all contributed to the summary recommendation, the main assessment that defined the overall decision was that of sub-PICO 6.1 on the comparison of the BPaLM regimen of the stage 2 (corresponds to Phase 3) in the TB-PRACTECAL trial with the mix of SoC regimens (conforming to the WHO-recommended 9-month or longer regimens). Even though the TB-PRACTECAL trial was not designed to compare the investigational regimens against each other and with the SoC, the comparisons of the different arms of the trial with the BPaLM arm (sub-PICOs 6.2–6.6) were performed to aid the panel in making final decisions.
The assessment of PICO 3 allowed for the decision on the optimal dosing and duration of linezolid within the BPaLM/BPaL regimen and narrowed down the subsequent comparisons to the intervention regimen with this particular dose and duration of linezolid – BPaL (600 mg – 26 weeks). The justification for how the other assessments have contributed to the overall recommendation can be summarized as follows:
- The assessment of PICO 4 resulted in the conditional recommendation for use of BPaL (600 mg – 26 weeks) regimen over the currently recommended longer regimens in patients with MDR/RR-TB and additional fluoroquinolone resistance.
- The three assessments performed under PICO 5 resulted in the conditional recommendations for the BPaL (600 mg – 26 weeks) regimen over the currently recommended 9-month regimen with ethionamide (sub-PICO 5.1), over longer regimens (sub-PICO 5.2) and over the new 9-month regimen where ethionamide is replaced with 2 months of linezolid (sub-PICO 5.3) in patients with pulmonary MDR/RR-TB without fluoroquinolone resistance.
- The assessment of sub-PICO 6.1 resulted in the conditional recommendation for use of the BPaLM regimen of the TB-PRACTECAL trial over the comparator, the mix of SoC regimens under this trial conforming to the WHO recommendations on 9-month or longer regimens, depending on the trial site.
- The assessments of sub-PICOs 6.4 and 6.6 resulted in the conditional recommendations for BPaLC and BPaL over the SoC in the TB-PRACTECAL trial; thus all three 6-month BPaL-based regimens were assessed to be preferred over the mix of SoC regimens under this trial.
- The assessments of sub-PICOs 6.3 and 6.5 resulted in the conditional recommendations for BPaLM and BPaL over BPaLC; based on these assessments the GDG concluded that BPaLC should not be recommended as a regimen.
- The assessment of sub-PICO 6.2 resulted in the conditional recommendations for BPaLM over BPaL; thus, it highlighted the use of the BPaLM regimen as the preferred regimen under the conditions specified in the recommendation and remarks. Compared with BPaL, BPaLM led to more treatment success, fewer failures or recurrences and less emerging drug resistance while showing little difference in AEs.
Table 1.3. PICO questions and decisions of the GDG panel
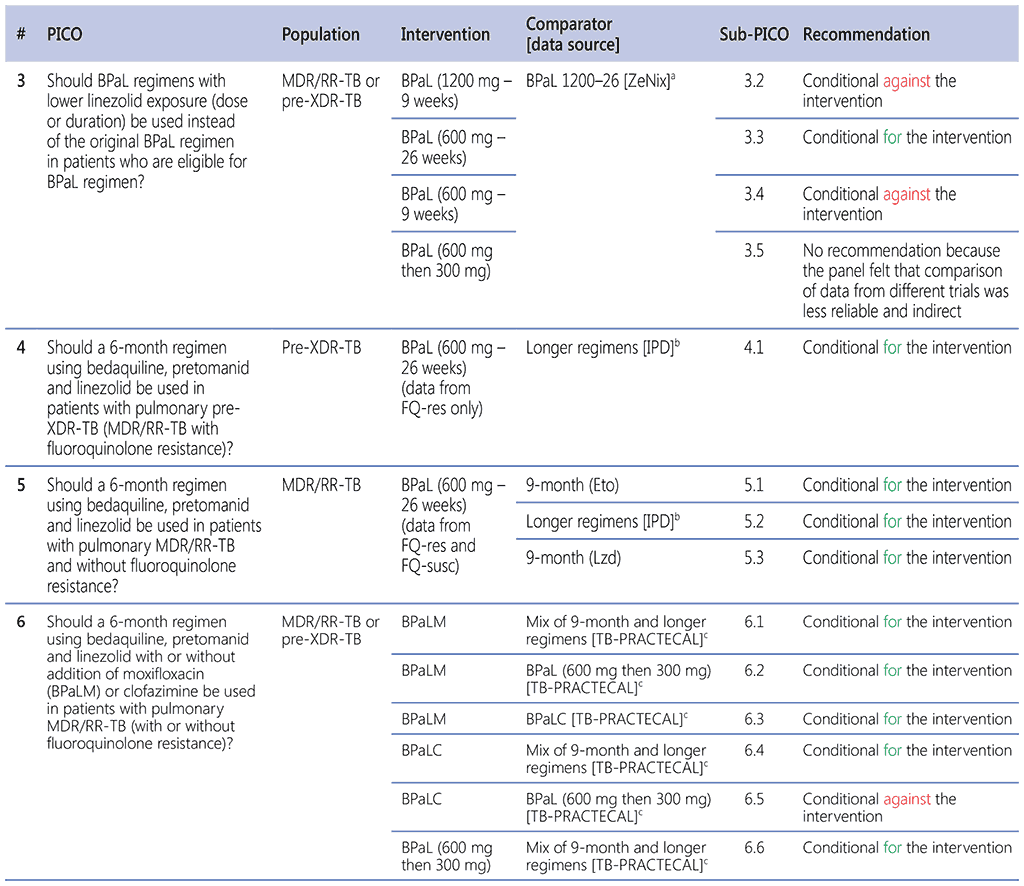
BPaL: bedaquiline, pretomanid and linezolid; BPaLC: bedaquiline, pretomanid, linezolid and clofazimine; BPaLM: bedaquiline, pretomanid, linezolid and moxifloxacin; Eto: ethionamide; FQ-res: fluoroquinolone resistant; FQ-susc: fluoroquinolone susceptible; GDG: Guideline Development Group; IPD: individual patient data; Lzd: linezolid; MDR/RR-TB: multidrug-resistant or rifampicin-resistant TB; PICO: population, intervention, comparator and outcome; TB: tuberculosis; pre-XDR-TB: pre-extensively drug-resistant TB.
a ZeNix trial.
b 2021 IPD.
c TB-PRACTECAL.
The GDG panel discussed the subgroups and implementation considerations, and the monitoring and evaluation and research priorities as they pertain to the summary recommendation rather than for each individual sub-PICO question.
Subgroup considerations
Children and adolescents
Children were excluded from the ZeNix trial (aged 0–13 years) and the TB-PRACTECAL trial (aged 0–14 years); therefore, no analysis specific to this subgroup of patients could be performed. All medicines in the BPaLM regimen have been used in children except for pretomanid. New data on bedaquiline has been recently reviewed and its use has been expanded to all ages (see additional recommendation in the section on longer regimens and (30)). The lack of safety data on pretomanid in children aged below 14 years was the main barrier for potential extrapolation of the BPaLM/BPaL recommendation to the threshold of being aged below 14 years. Thus, the recommendation of the BPaLM/BPaL regimen applies to adults and adolescents aged 14 years and older.
People living with HIV
HIV was diagnosed in 34 of 172 (19.8%) people enrolled in the ZeNix trial; however, it was impossible to perform any adjusted stratified analyses for people living with HIV (PLHIV), owing to the small sample size in sub-PICO comparisons 3.2, 3.3, 3.4 and 3.5. PLHIV were eligible for enrolment in the ZeNix trial if they had a CD4 count of more than 100 cells/mm³ and if they were using antiretroviral medications.³¹ No aspects specific to HIV status or CD4 count were in the list of TB-PRACTECAL exclusion criteria, and PLHIV represented 27% of those enrolled. The median CD4 count among PLHIV was 322 (interquartile range [IQR] 217–622) across the four arms.
It is important to take drug–drug interactions into account when administering TB and HIV medications in combination; such interactions are discussed below under implementation considerations. Although some therapies are to be avoided, there are alternative antiretroviral agents that can be considered when pretomanid is used. Thus, the recommendation of the BPaLM/BPaL regimen applies to all people regardless of HIV status, although some caution should be used when enrolling patients with CD4 counts lower than 100 cells/mm³.
Pregnant and breastfeeding women
Pregnant and breastfeeding women were excluded from the ZeNix and TB-PRACTECAL trials owing to unknown effects of the new medicine, pretomanid, on fetal development; therefore, no analysis specific to this subgroup of patients could be performed. The use of bedaquiline in pregnancy has been associated with infants born with a lower mean birth weight than infants whose mothers did not take bedaquiline; however, when infants were followed up over time, no evidence of late adverse impacts was found (see Treatment of drug-resistant TB using longer regimens). Breastfeeding is not recommended for women taking pretomanid (39). Thus, the recommendation of the BPaLM/BPaL regimen does not apply to pregnant and breastfeeding women. While the safety of pretomanid during pregnancy and breastfeeding is unclear, other treatment options need to be used.
Extrapulmonary TB
Patients with extrapulmonary TB were excluded from the ZeNix and TB-PRACTECAL trials; therefore, no analysis specific to this subgroup of patients could be performed. The available data on the central nervous system (CNS) penetration of bedaquiline or pretomanid are limited. Although all forms of extrapulmonary TB were excluded from the clinical trials, the GDG felt that extrapolation to extrapulmonary TB and other forms of TB was warranted except in cases involving severe forms of TB that may require special treatment arrangements and decisions, particularly TB involving the CNS, osteoarticular and disseminated forms of TB. Thus, the recommendation of the BPaLM/BPaL regimen applies to people with pulmonary TB and all forms of extrapulmonary TB except for TB involving the CNS, and osteoarticular and disseminated forms of TB.
Implementation considerations
High treatment success rates shown for the BPaLM and BPaL regimens in the Nix-TB study and in the ZeNix and TB-PRACTECAL trials, and favourable comparison with the current SoC regimens led to thorough discussions during the GDG meeting of an overall recommendation for implementation under routine programmatic conditions and of the implementation considerations for this regimen. Given that this recommendation is conditional, the results of additional or ongoing operational research will help to add further knowledge that can be used to adjust and improve implementation guidance for the regimen.
Patient selection
Overall, to reproduce the treatment success rates observed in the ZeNix and TB-PRACTECAL trials, it is important to carefully select eligible patients. Once those patients are enrolled, it is also important to provide effective patient support to enable adherence to treatment. It is also important to maintain close monitoring for AEs, response to treatment and emerging drug resistance, and to properly manage adverse drug reactions and prevent complications from drug–drug interactions.
The selection of patients is best aligned with the eligibility criteria of two trials (also reflected in the subgroup consideration above). The patients that can be enrolled on the BPaLM/BPaL regimen should have bacteriologically confirmed MDR/RR-TB, with or without resistance to fluoroquinolones.
Drug susceptibility testing
It is important to pay attention to the previous use and susceptibility status of the medicines comprising this regimen. Patients with a known history of more than 1 month use of bedaquiline, pretomanid (or delamanid, given some degree of cross-resistance) and linezolid should not be enrolled on this regimen, unless the results of recent DST of these medicines has confirmed susceptibility. In cases where there is no prior use of these medicines or confirmed susceptibility, fluoroquinolone resistance testing should also be done before the start of treatment. However, fluoroquinolone resistance testing should not delay treatment initiation (e.g. in cases where this DST is not available or results are delayed). When DST results confirm fluoroquinolone susceptibility, treatment can be continued without any modifications. In cases of fluoroquinolone resistance, moxifloxacin should be dropped and the regimen continued as the BPaL combination only. This modification may seem counterintuitive, because patients with TB that is resistant to an increased number of drugs will receive fewer TB medicines. However, moxifloxacin is unlikely to provide a benefit in the presence of fluoroquinolone resistance and the BPaL regimen has been shown to have high efficacy without moxifloxacin. In the context of fluoroquinolone resistance, omitting moxifloxacin will help to avoid potential toxicity related to this medicine. Conversely, in the absence of fluoroquinolone resistance, the use of moxifloxacin further increases the efficacy of the regimen and may provide protection against acquired bedaquiline resistance, and thus is recommended. If fluoroquinolone DST results are unavailable, the GDG judged the likely benefits of retaining moxifloxacin as part of the regimen as outweighing the potential harms; therefore, WHO suggests using the BPaLM regimen in this situation.
The establishment and strengthening of DST services is a vital consideration for implementation of all treatment regimens for MDR/RR-TB. In patients with bacteriologically confirmed MDR/RR-TB, the Xpert® MTB/XDR (Cepheid) or GenoType® MTBDRsl (Hain Lifescience) assays may be used as the initial test, in place of culture and phenotypic DST, to detect resistance to fluoroquinolones (40, 41). The WHO has recently endorsed the use of targeted next-generation sequencing (NGS) for the detection of drug resistance, as outlined in the “WHO consolidated guidelines on tuberculosis. Module 3: Diagnosis – rapid diagnostics for tuberculosis detection, third edition” (Geneva: World Health Organization; 2024). Targeted NGS involves amplifying specific genes and then using sequencing technology to identify resistance markers to multiple drugs in one test. This method can scrutinize entire genes for mutations linked to resistance, potentially offering greater accuracy than other molecular tests recommended by WHO. It is particularly useful for detecting resistance to three drugs in the BPaLM regimen – bedaquiline, linezolid, and moxifloxacin. For pretomanid, the fourth drug in this regimen, new drug susceptibility testing (DST) criteria have been set. Countries implementing programs to manage drugresistant TB (DR-TB) should develop lab infrastructure to perform culture-based phenotypic DST for drugs used in multidrug-resistant/rifampicin-resistant TB (MDR/RR-TB) treatments, where reliable methods exist (e.g., bedaquiline, linezolid, pretomanid, cycloserine, clofazimine, and delamanid). Additionally, the latest guidelines include a new molecular test for pyrazinamide, which should simplify its testing process (42).
Currently, there is limited capacity globally for DST for bedaquiline and linezolid. As these medicines and regimens containing these medicines become more widely used, laboratory capacity in this area must be strengthened. National and reference laboratories will need to have necessary facilities and reagents to make DST available; also, they will need data on the minimum inhibitory concentration (MIC) distribution of all M. tuberculosis lineages that are circulating globally. Establishing or expanding capacity for sequencing of M. tuberculosis can provide a strong and future-proof platform for DST. If resistance to any of the component medicines in the BPaL regimen is detected, treatment with another recommended regimen should be started. The WHO TB Supranational Reference Laboratory (SRL) Network is available to support national TB reference laboratories in performing quality-assured DST. A WHO technical consultation in established critical concentrations for DST for the fluoroquinolones, bedaquiline, delamanid, clofazimine, pretomanid and linezolid (42, 43). When methods for DST are available, countries will need to add surveillance of resistance to new medicines to their routine efforts or surveys. These data can guide the adoption and use of new regimens and can also protect against amplification of resistance profiles.
Drug–drug interactions
It is important to take drug–drug interactions into account when administering TB and HIV medications in combination, including the documented interactions between bedaquiline and efavirenz (44). Efavirenz reduces pretomanid exposures significantly; therefore, an alternative antiretroviral agent should be considered if pretomanid forms part of the BPaLM/BPaL regimen (39). The preferred ART regimens for co-administration with BPaLM/BPaL are dolutegravir-based regimens in combination with two nucleoside reverse transcriptase inhibitors.
Other considerations
Several other groups of patients were excluded from the two trials; for example, patients with liver enzyme measurements three or more times over the upper limit of normal; people with a corrected QT interval by Fredericia (QTcF) more than 500 ms, or history of cardiac disease, syncopal episodes, significant arrythmias, congenital QT prolongation, torsade de pointes or cardiomyopathy; those with a current peripheral neuropathy of Grade 3–4; and moribund patients with very low BMI (<17). These groups of patients may only receive the regimen if the treating physician judges this to be the best option despite these contraindications and where adequate monitoring is available.
Regimen composition, dosing of component medicines and frequency
The BPaLM/BPaL regimen consists of bedaquiline, pretomanid and linezolid, with or without moxifloxacin throughout the regimen duration. Pretomanid is administered at 200 mg once daily for the duration of the regimen. When moxifloxacin is part of the regimen, it is dosed at 400 mg once daily throughout the treatment course. The fluoroquinolone of choice used in the TB-PRACTECAL trial was moxifloxacin; given that no evidence on using other fluoroquinolones was available at the time of the GDG assessments, the replacement of moxifloxacin with levofloxacin or any other fluoroquinolone cannot be recommended at this stage. The frequency of dosing should be 7 days a week with treatment support or using video-supported therapy; that is, as it was administered in both the trials.
Bedaquiline dosing schemes
The TB-PRACTECAL and ZeNix trials used slightly different dosing schemes for bedaquiline although the overall drug exposure was comparable (31). The dosing schedule used in the TB-PRACTECAL trial was consistent with the product label whereas the dosing schedule used in the ZeNix trial presented the advantage of daily dosing throughout the regimen and may be used as one of the options for administration. Either of the bedaquiline dosing schemes may be used for programmatic implementation:
- daily throughout treatment: 200 mg once daily for 8 weeks followed by 100 mg once daily; and
- daily for loading dose and three times per week thereafter: 400 mg once daily for 2 weeks followed by 200 mg three times per week.
Dosing of linezolid
The ZeNix trial used several different dosing and duration schemes of linezolid, with the aim of determining the optimal administration schedule for this medicine. Linezolid is known to cause several potentially serious adverse effects; among those of most concern are peripheral neuropathy, optic neuritis and myelosuppression (45). The GDG review of the ZeNix trial data identified the optimal dosing for linezolid to be 600 mg once a day for 26 weeks, and this arm of the ZeNix trial was used for the main comparisons. Study participants in this arm of the trial received 600 mg of linezolid once daily for 26 weeks, with a reduction to 300 mg daily allowed in the event of linezolid specific toxicities. In the TB-PRACTECAL trial, dosing of linezolid was slightly different – participants were given 600 mg daily for 16 weeks and then 300 mg daily for the remaining 8 weeks (the duration of BPaLM in this trial was 24 weeks).
The GDG panel considered that it would be preferable to use linezolid 600 mg/daily throughout the regimen, but the dose can be reduced to 300 mg/daily if necessary to mitigate toxicity.
Regimen duration, changes and extensions
The BPaLM and BPaL regimens have been studied as the standardized courses of treatment. Therefore, modification of the regimen through early discontinuation or replacement of any of the component medicines may result in different (and possibly worse) treatment outcomes. In the TB-PRACTECAL trial, patients received 24 weeks of BPaLM. In the ZeNix trial, treatment was extended to a total of 9 months in patients on the BPaL regimen who remained sputum culture positive or who reverted to being sputum culture positive between months 4 and 6, or whose clinical condition suggested they may have progressive TB. In cases where treatment was interrupted and treatment duration was extended to make up for missed doses, it was necessary for patients to complete 6 months of the regimen (i.e. 26 weeks of prescribed doses) within 8 months; also, for patients in whom treatment was extended, it was necessary to complete 9 months of treatment (i.e. 39 weeks of prescribed doses) within 12 months.
Eligible patients with susceptibility to fluoroquinolones can be started on the BPaLM regimen for 6 months, with dosing of individual medicines as described above. This combination of medicines can be continued throughout the regimen without any prolongation (unless there is a need to make up the missed doses). In cases where resistance to fluoroquinolones is identified before or after treatment initiation, moxifloxacin can be discontinued. When the regimen is BPaL from the start or is changed to BPaL, it can be extended to a total of 39 weeks (counting from the start of the therapy with BPaLM/BPaL). This extension is justified in cases of failure to convert culture between months 4 and 6 while on treatment; alternatively, it can be based on the clinical judgement of the treating physician. Up to 1 month can be added to the overall treatment duration if there is a need to make up the missed doses.
The GDG panel acknowledged these slight differences in the treatment duration of the BPaLM and BPaL regimens as studied in these two trials, and suggested standardizing the treatment duration of BPaLM to 6 months (26 weeks) during programmatic implementation; for BPaL they suggested the possibility of extension to a total of 9 months (39 weeks) if sputum cultures are positive between months 4 and 6. All medicines in the regimen are to be used throughout treatment duration, including a potential extension from 26 to 39 weeks (when BPaL is used). Ideally, missing doses of all three or four drugs in the regimen should be avoided; however, if doses are missed, any interruption of longer than 7 days should be made up by extending the treatment duration (for the number of missed doses); therefore, 26 or 39 weeks of prescribed doses should be completed within an overall period of 7 or 10 months, respectively.
Missing doses and tolerances for treatment interruptions
The TB-PRACTECAL and ZeNix trials used different tolerances for treatment interruption and missing doses, and the ZeNix trial protocol provided specific rules for linezolid administration.
The GDG panel suggested standardizing the allowable missing doses and the approach to linezolid administration. The following pragmatic approach is suggested to guide clinical judgement and potential minor deviations in individual cases:
- all possible efforts should be made to support the patient and manage the AEs to ensure uninterrupted treatment and intake of all medicines in the regimen; however, when medicine cannot be tolerated it should be stopped;
- consecutive treatment interruption (of all medicines in the regimen) of up to 2 weeks should be made up and added to the treatment duration;
- non-consecutive missed doses of all medicines in the regimen up to a cumulative total of 4 weeks should be made up and added to the treatment duration; and
- after consecutive administration of linezolid at recommended doses (600 mg/daily) for at least 9 weeks, in case of intolerability the dose can either be adjusted down (to linezolid 300 mg/daily) or omitted (while other medicines in the regimen are continued) for a total of a maximum of 8 weeks throughout the treatment course.
In case any single one of these tolerances is exceeded, a thorough assessment of the patient’s status will be required to decide whether to continue the treatment strategy or modify it.
Recommendation 1.2 The 6-month bedaquiline, delamanid, linezolid, levofloxacin and clofazimine (BDLLfxC) regimen

Remarks
- Drug susceptibility testing (DST) for fluoroquinolones is strongly encouraged in people with MDR/RR-TB. Although it should not delay the initiation of the BDLLfxC, the test results should guide the decision on whether levofloxacin or clofazimine should be retained.
- This recommendation applies to the following:
- People with MDR/RR-TB or pre-XDR-TB (i.e. MDR/RR-TB and resistance to fluoroquinolones)
- Patients with less than 1 month of previous exposure to bedaquiline, linezolid, delamanid or clofazimine. When exposure is greater than 1 month, these patients may still receive the regimen if resistance to the specific medicines involved in such exposure has been ruled out.
- People with diagnosed pulmonary TB of all ages, including children, adolescents, PLHIV, and pregnant and breastfeeding women.
- People with all forms of extrapulmonary TB except for TB involving the CNS, osteoarticular TB, or disseminated forms of TB with multiorgan involvement.
- Children and adolescents who do not have bacteriological confirmation of TB or resistance patterns but who do have a high likelihood of MDR/RR-TB (based on clinical signs and symptoms of TB, in combination with a history of contact with a patient with confirmed MDR/RR-TB).
- When resistance to fluoroquinolones is unknown, the regimen should be started as BDLLfxC and then adjusted based on the DST results. In cases of quinolone susceptibility, clofazimine should be dropped and the regimen started or continued as bedaquiline, delamanid, linezolid and levofloxacin (BDLLfx). In cases of resistance to fluoroquinolones, levofloxacin should be dropped and the regimen started or continued as bedaquiline, delamanid, linezolid and clofazimine (BDLC).
Rationale
The rationale for this recommendation is based on the evidence and considerations described in detail in the following two subsections. Data from an RCT (BEAT Tuberculosis trial: NCT04062201) showed that the efficacy and safety of the BDLLfxC regimen was similar to that of the control arm regimen (WHO-recommended 9-month or longer regimens), and offers advantages based on its shorter duration and lower pill burden than the comparators. It was judged that implementing this regimen was probably feasible and acceptable, with equity probably improved, and that implementing this regimen was probably suitable for most population groups, including pregnant and breastfeeding women, children and PLHIV.
Summary of evidence
This section provides the PICO question, the data and studies considered to answer the questions, the methods used for analysis and data synthesis, a summary of evidence on desirable and undesirable effects and the certainty of that evidence, and a summary of other evidence considered during the recommendation’s development. Additional details on the evidence are available in the web annex containing the GRADE evidence summary tables and GRADE EtD tables (Annex 5).
PICO question
The recommendation in this section is a result of assessments of the PICO question below:
PICO question 1–2024 (MDR/RR-TB, 2024): Should a 6-month regimen using bedaquiline, delamanid and linezolid – with or without the addition of levofloxacin or clofazimine or both (BDLLfxC) – be used in patients with pulmonary RR-TB (with or without fluoroquinolone resistance) over the currently recommended 9-month regimen?
Data and studies considered
The review of this group of PICO questions during the GDG meeting convened by WHO in June 2024 was based on the new evidence from the BEAT-TB trial in South Africa. That trial was an RCT led by the University of the Witwatersrand, designed to compare the safety and efficacy of a novel regimen, BDLLfxC, for the treatment of MDR/RR-TB or pre-XDR-TB with either a 9-month oral regimen (linezolid containing) or a longer individualized regimen. Patient populations included in this trial were recruited based on the criteria shown in Table 1.4.
Table 1.4. High-level summary of main inclusion and exclusion criteria: BEAT-TB trial

CNS: central nervous system; ECG: electrocardiogram; FQ: fluoroquinolone; HIV: human immunodeficiency virus; RR-TB: rifampicin-resistant TB; P: QTcF: corrected QT interval by Fredericia formula ; TB: tuberculosis; UNL: upper normal limit.
BEAT Tuberculosis trial
This was an open-label, Phase 3, non-inferiority RCT designed to establish the efficacy and safety of a regimen comprising 6 months (24 weeks) of bedaquiline (B), delamanid (D), and linezolid (L), with levofloxacin (Lfx) and clofazimine (C) compared with the current South African SoC for the treatment of MDR/RR-TB or pre-XDR TB.
The objectives of the trial were to compare the proportion of participants with a successful outcome at the end of treatment and the end of follow-up at 76 weeks after randomization on the study regimen with the proportion with a successful outcome on the control regimen, and to compare the proportion of participants who experienced grade 3 or more significant AEs during treatment.
Eligible participants between 6 and 12 years diagnosed with confirmed or probable pulmonary RR-TB and aged above 12 years with confirmed RR-TB were randomized to receive either the experimental or SoC regimen. All participants were followed up for 76 weeks from randomization.
The trial used a treatment strategy in which either levofloxacin or clofazimine was dropped from the regimen depending on FQ DST results: BDLLfxC initiated without delay in the case of unknown FQ-resistance at the time of RR-TB diagnosis (and continued with both levofloxacin and clofazimine if FQ-DST results could not be obtained); or BDLLfx continued for FQ-susceptible TB; or BDLC for FQ-resistant TB. Within the trial, outcomes with this treatment strategy were compared with outcomes with the recommended all-oral, bedaquiline-containing 9-month or longer regimens (most of the control group received a 9-month linezolid-containing regimen). The dataset included patients with severe TB disease, PLHIV, children aged 8 and above, adolescents and a small group of pregnant and breastfeeding women. The regimen could be given for 6 months or extended to 9 months if there was no clinical or bacteriological improvement.
The control group included participants on the WHO-recommended 9-month regimen with 2 months of linezolid (the SoC in South Africa at the time), and fewer patients on the longer individualized regimens designed for patients with pre-XDR-TB.
Methods used for analysis and data synthesis
Descriptive analyses of the baseline characteristics of participants in the study were performed. Characteristics included demographics, pregnancy status, laboratory parameters (e.g. HIV status and CD4 count, if applicable), drug susceptibility tests and diagnostic test results, TB treatment received before randomization, AEs and treatment regimens, and end-of-treatment and end-of-follow-up outcomes.
Statistical analyses were based on the WHO treatment outcome definitions listed in Annex 2, slightly modified for use in a clinical trial that included post-treatment follow-up to 76 weeks after randomization. The study team performed the analyses and presented them to the WHO panel. For the end of follow-up outcome measured at 76 weeks after initiation of treatment, a successful outcome was defined as follows:
- Cured – culture negative at the end of follow-up or culture negative when last seen, if the participant was lost before the end of follow-up and provided they had a successful treatment outcome at the last study visit attended.
For the end-of-follow-up outcome measured at 76 weeks after initiation of treatment, an unsuccessful outcome was defined as one of the following:
- Recurrence – two consecutive positive cultures separated by at least 14 days, or one positive culture after confirmed culture conversion with clinical signs and symptoms of TB, or no improvement or worsening of radiological changes since baseline. An isolated positive smear or culture without clinical or radiographic deterioration after treatment completion provides insufficient evidence to define recurrent TB.
- Death (from any cause) during follow-up.
- Loss to follow-up with clinical signs or symptoms of TB (or both) when last seen, or sputum culture positive when last seen, or not sputum culture negative and with clinical signs and symptoms of TB when last seen.
For the primary analysis, a successful outcome was required at the end of treatment and at the end of follow-up.
If treatment was modified or extended for participants allocated to either strategy, those people may still have been on treatment at the final follow-up visit 76 weeks after randomization. In this situation, for the primary efficacy outcome classification, the end of treatment outcome was defined at this time point (or the last participant visit while on treatment) and the end of follow-up outcome was considered to be the same as the end of treatment outcome.
The primary safety endpoint was the incidence of all treatment-emergent AEs of Grade 3 or higher, using the Division of AIDS (DAIDS) table for grading the severity of adult and paediatric AEs by treatment arm and up to 30 days following the end of treatment. Serious AEs were collected until the end of the follow-up.
Table 1.5. Cross-tabulation of outcomes from the BEAT-TB trial and WHO
LTFU: loss to follow-up; TB: tuberculosis; WHO: World Health Organization.
a Culture-negative at week 76 or cured at the end of treatment, culture-negative when last seen with no symptoms of TB.
b Three participants experienced treatment failure and then subsequently died.
Decision thresholds
This recommendation was developed using a new method for determining the magnitude of the health effects. A triangulation approach was used to develop outcome-specific decision thresholds (DTs) for judging the magnitude of the effects for the following health outcomes: death, sustained treatment success, treatment failure or recurrence, LTFU, AEs and amplification of drug resistance. GDG members deemed these outcomes critical or important for decision-making based on a prioritization survey using the GRADE approach. The survey included health outcome descriptors that had previously been developed for each of the health outcomes to facilitate understanding of the outcomes by the GDG members during their decision-making process.
The GDG first reviewed judgements about the magnitude of health effects made by other GDGs for prior WHO MDR-TB guidelines (3, 14), to determine approximate ranges of effect sizes that the group considered to be trivial, small, moderate or large. Members then identified a systematic review to help inform suggested health utility values for the health state of having DR-TB disease and treatment success (reported as about 0.5 and 0.9, respectively) (46). For the other outcomes (treatment failure, LTFU, amplification of drug resistance and AEs), a health utility value of 0.5 was used, considering that these would be similar health states to having DR-TB disease, and to align with previous judgements made in other TB guidelines.
The group then used the empirical evidence from the GRADE THRESHOLD trial (47) to calculate suggested utility-adjusted absolute effect thresholds for the health outcomes. The calculated thresholds were as follows (48):
- death (health utility: 0):
- trivial or no effect: ≤14 fewer or more deaths per 1000 people;
- small effect: 15–32 fewer or more deaths per 1000 people;
- moderate effect: 33–63 fewer or more deaths per 1000 people;
- large effect: ≥64 fewer or more deaths per 1000 people;
- sustained treatment success (health utility: 0.9):
- trivial or no effect: ≤15 fewer or more treatment successes per 1000 people;
- small effect: 16–35 fewer or more treatment successes per 1000 people;
- moderate effect: 36–68 fewer or more treatment successes per 1000 people;
- large effect: ≥69 fewer or more treatment successes per 1000 people;
- treatment failure or recurrence, LTFU, AEs and amplification (acquisition) of drug resistance (all with health utility 0.5):
- trivial or no effect: ≤30 fewer or more failures or recurrences per 1000 people;
- small effect: 31–59 fewer or more failures or recurrences per 1000 people;
- moderate effect: 60–119 fewer or more failures or recurrences per 1000 people; and
- large effect: ≥120 fewer or more failures or recurrences per 1000 people.
These suggested thresholds (assumed to occur over the duration of follow-up in the trials) were generally consistent with judgements that were made in the previous WHO MDR-TB guidelines (3).
Finally, in preparation for the GDG meeting at which recommendations would be formulated, an online survey was administered to the group to obtain their feedback on the suggested DTs. The survey asked members to agree with the suggested thresholds, or to disagree and suggest alternative thresholds based on their expert experience. The agreed-upon thresholds were again reviewed at the start of the GDG meeting, and the group decided to use those thresholds to inform their judgements about magnitude of health effects in the GRADE EtD frameworks. Figures were created to visually depict absolute effects and 95% confidence intervals (CIs) from the research evidence of the relevant trial in relation to the DTs for each health outcome, to facilitate the GDG’s discussion and judgements about whether the health effects were trivial, small, moderate or large, and to judge the level of imprecision of estimates (Fig. A1.3). The same thresholds were used to inform the group’s judgements about imprecision, in line with GRADE guidance for decision-making (49).
Summary of evidence on desirable and undesirable effects and certainty of evidence
Outcomes among patients with MDR/RR-TB with or without quinolone resistance (MDR/RR-TB or preXDR-TB) receiving the BDLLfxC regimen were compared to those receiving the SoC (9–12-month) all-oral regimens with linezolid for patients with MDR/RR-TB; 18–20 month all-oral regimens for patients with pre-XDR-TB).
Participants with MDR/RR-TB (with or without quinolone resistance) receiving the BDLLfxC regimen (n=202) compared to participants receiving WHO-recommended SoC regimens used in the BEAT-TB trial (n=200) experienced:
- higher levels of failure or recurrence: 8.4% vs 7.0%; RD=14 more per 1000, 95% CI: 38 fewer to 66 more per 1000)
- lower levels of death: 5.0% vs 5.0%; RD= 0.5 fewer per 1000 (95% CI: from 43 fewer to 42 more per 1000);
- lower levels of LTFU: 1.0% vs 2.0%; RD= 10 fewer per 1000 (95% CI: from 34 fewer to 14 more per 1000);
- lower levels of grade 3–5 AEs: 34% vs 38%; RD=38 fewer per 1000 (95% CI: 132 fewer to 55 more per 1000); and
- lower levels of amplified resistance: 2.5% vs 3.0%; RD=5 fewer per 1000 (95% CI: 37 fewer to 27 more per 1000).
The GDG also considered the duration and pill burden with the intervention and comparator regimens. The intervention regimen is 24 weeks (5.5 months), so treatment duration is reduced compared with the control arm by 3–18 months. The exact magnitude of reduction in time on treatment depends on the specific comparator regimen, which includes shorter (9–12 months) and longer (18–24 months) regimens. The pill burden of the intervention regimen is lower than that for the comparator regimens. The exact magnitude of reduction in pill burden depends on the specific comparator regimen.
The GDG judged the benefits of BDLLfxC to be small and the undesirable effects to be trivial compared with WHO-recommended SoC regimens. The overall certainty of evidence was very low, primarily due to imprecision in the effect estimates. Based on this, the panel determined that the balance of health effects probably favours BDLLfxC regimen.
Fig. 1.1. Visual depiction of absolute effects in relation to the decision thresholds used by the GDG
Note: The positioning of point estimates and CIs is indicative and for illustration purposes only; exact figures are available in the GRADE evidence profiles. As indicated above in the sections on desirable and undesirable effects, decision thresholds vary for some of the outcomes and therefore only the descriptive labels (trivial, small, moderate or large) are used for the x-axis rather than numerical values.
Summary of other evidence
Resources required and cost–effectiveness
Additional data reviewed by the GDG relevant to these PICO questions were the estimates of the regimen price provided by the Stop TB Partnership’s GDF, based on the drug pricing included in the GDF online catalogue (50) on 20 June 2024.
Table 1.6. Regimen cost estimates
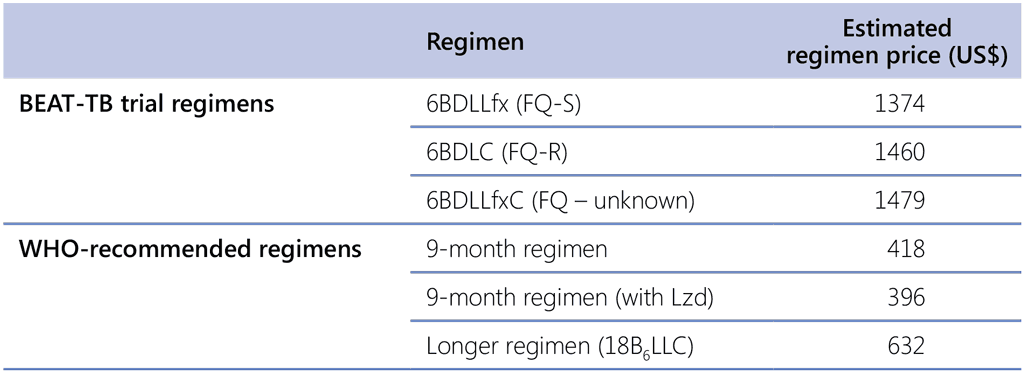
BDLLfx: bedaquiline, delamanid, linezolid and levofloxacin; FQ: fluoroquinolones; FQ-R: FQ resistant; FQ-S: FQ susceptible; Lzd: linezolid; WHO: World Health Organization.
The GDG also considered the following example of country-specific costs (both patient-borne and health system costs) over a 3-month span (excluding drug costs), based on modeling analysis in Ryckman et al. (2024) (51). Costs may vary depending on the composition of the regimen being used.
Table 1.7. Patient-borne and health system costs

RR-TB: rifampicin-resistant TB; TB: tuberculosis.
The GDG noted that affordability will vary depending on country (and resources available), health system differences and the population the regimen would be used for; accordingly, they judged that costs would vary between moderate and large.
The price of delamanid is one of the major cost drivers in BDLLfxC. The drug is off-patent; hence, prices may decrease with generic development. It was highlighted that there are costs to a longer duration of treatment (especially for patients and families but also the health system) – some estimates were available and discussed by the group. Considering these costs together with the drug prices may attenuate some of the increased costs for the health system and lead to cost savings from the patient perspective. However, countries looking to implement a specific treatment regimen typically focus on the drug cost. No evidence on cost–effectiveness was available.
Equity, acceptability and feasibility
When considering the impact on health equity, the panel considered the treatment duration and the ability to decentralize treatment (to enable access for remote, underserviced settings and disadvantaged populations).
The GDG highlighted that health equity would probably increase for particular parts of the population that would have access to the shorter regimen and the medications included (e.g. pregnant women and children, and possibly other groups as well). On the other hand, certain populations might not be able to afford the regimen, and there could be differences in impact on health equity among countries because of differences in availability of medications.
Despite not being able to identify relevant research evidence, the panel used their collective experience to judge that there would probably be advantages associated with the use of the BDLLfxC regimen owing to its reduced complexity and shorter duration. The panel judged that use of the BDLLfxC regimen would probably increase equity.
When considering the acceptability of the intervention, the panel considered patients and health care providers as key stakeholders. The GDG considered the following aspects as critical with regards to acceptability: regimen duration and drug safety monitoring needs (relating to necessary travel, loss of income and general disruption of the life of patients; and workload for the health care system), and needs for DST. The GDG highlighted that delamanid requires taking medicines twice per day in this regimen, which may affect acceptability. The panel judged that the BDLLfxC regimen would probably be acceptable.
The GDG considered the following aspects to affect feasibility (i.e. to be potential barriers to implementation): regulatory approval of drugs in the regimen, requirements for drug safety monitoring and requirements for DST.
BEAT-TB was a pragmatic trial. Similar intervention regimens have been given in other studies across a range of countries, increasing the likelihood that the implementation of BDLLfxC is feasible in settings beyond the trial setting in South Africa.
Approval by regulators influences the access and feasibility of implementing the regimen; also, alternative regimens are not always available. Access to some of the medications is hampered by licensing differences. For example, in the USA, there is no Food and Drug Administration (FDA) approval for delamanid, it is only available under compassionate use restrictions.
A few patients with low hemoglobin levels in the BEAT-TB trial received a blood transfusion before initiation of treatment; the need for this may be a limiting factor in some settings for patients with low hemoglobin levels.
Overall, given the reduced duration, complexity and associated workload, the panel judged that implementation is probably feasible.
Conclusion
The BDLLfxC regimen is suggested over currently recommended 9-month or longer regimens in patients with MDR/RR TB.
Evidence to recommendations: considerations
Based on the decisions taken during the review and the combination of assessments described above, the new recommendation is to use the BDLLfxC regimen rather than the 9-month or longer (18-month) regimens in MDR/RR-TB patients. The BDLLfxC regimen is intended for use under routine programmatic conditions, which include pregnant and breastfeeding women and children aged below 14 years. However, the cost of the regimen (driven by the price of delamanid) remains a barrier to widespread adoption.
Subgroup considerations
Based on research evidence and expert experience, the panel identified subpopulations of people who might be affected differently than most by this recommendation; these subpopulations were PLHIV, children, pregnant and breastfeeding women, patients with extrapulmonary TB and patients with extensive TB disease and resistance to fluoroquinolones. The recent new recommendation for the use of bedaquiline in children with MDR/RR-TB aged below 6 years was considered (30). The panel noted specific considerations for the subpopulations listed below.
Children and adolescents
Thirty participants aged 8–17 years with confirmed RR-TB were enrolled in the study, with 17 in the control group and 13 in the intervention group. Among them, seven (24%) had FQ resistance. One participant aged 17 years was found to have resistance to bedaquiline and clofazimine at baseline and was switched to an individualized rescue regimen. There were three Grade 3 or 4 AEs related to linezolid (1 case each of anemia, peripheral neuropathy and optic neuritis). Delamanid was discontinued in one participant on the control arm owing to neuropsychiatric side effects.
In addition to the trial data, all medicines in the BDLLfxC regimen have been used in children and adolescents, have well-documented safety and efficacy profiles, and have sufficient pharmacokinetic and pharmacodynamic data. Thus, the recommendation of the BDLLfxC regimen applies to adults, adolescents and children of any age.
People living with HIV
All participants of the BEAT-TB trial were required to undergo an HIV test, but the test result and their CD4+ count, or whether they were receiving ART at the time of randomization, did not affect their eligibility for the trial. The management of HIV infection followed the guidelines of the National Department of Health of South Africa. ART included either dolutegravir or a protease inhibitor to prevent drug–drug interactions, and non-nucleoside reverse transcriptase inhibitors were prohibited. In BEAT-TB, 202 PLHIV were enrolled, with 105 randomized to the study arm. The median CD4+ in this group was 168 (95% CI: 85.0, 299). No major effect of HIV status on the efficacy of the intervention regimen vs the control was observed (risk difference 2.1% (95% CI: –7.9%, +12.2%). Thus, the recommendation for the BDLLfxC regimen applies to all people, regardless of HIV status or degree of immunosuppression and appeared preferable to the comparator.
Pregnant and breastfeeding women
The components of BDLLfxC have been used in pregnancy in women with MDR/RR-TB. Pregnant women could be enrolled in the BEAT-TB trial in any trimester of pregnancy. Also, women who become pregnant during the trial could continue their treatment without any changes. Of the 10 pregnant women (4 to the trial regimen) enrolled, all pregnancies resulted in singleton live births, with one premature delivery. One woman in the study strategy experienced a relapse. There were two severe AEs in this group, but neither were drug-related.
Based on existing experience using the BDLLfxC regimen’s component drugs, the trial data and other currently recommended alternatives, the BDLLfxC regimen is recommended for use in pregnant and breastfeeding women.
Extrapulmonary TB
Patients with complicated extrapulmonary TB involving the CNS, osteoarticular and pericardial forms of TB were not included in the BEAT-TB trial. Therefore, no specific analysis could be performed for this patient subgroup. Patients with uncomplicated extrapulmonary TB were included if they also had pulmonary TB. Thus, the recommendation for the BDLLfxC regimen applies to people with pulmonary TB and all forms of extrapulmonary TB except for TB involving the CNS, osteoarticular or disseminated forms of TB with multiorgan involvement.
Extensive TB disease and resistance to fluoroquinolones
Patients with DR-TB who have extensive disease face greater challenges in treatment across all regimens and are at a higher risk of unfavourable outcomes and amplification of drug resistance. This risk is further elevated in those infected with pathogens that have advanced resistance profiles, particularly with additional resistance to fluoroquinolones. These patients may experience slightly worse outcomes compared to those without pre-XDR and develop resistance to regimen components. In the BEAT-TB trial among patients with pre-XDR-TB, the outcomes also appeared worse than among those who were fluoroquinolone susceptible. This reduction in the proportion with successful treatment outcomes was numerically greater in the BDLLfxC arm than in the control arm (14.3% vs 3.3%); however, this difference was within the range of statistical uncertainty and the GDG judged that this should not affect the recommendation or remarks. The rate of acquisition of resistance to bedaquiline was similar between the two groups (11% in BDLLfxC and 9.1% in control). The evidence from the BEAT-TB trial is insufficient to draw definitive conclusions due to the small sample when stratified by fluoroquinolone resistance and related imprecision. Nonetheless, clinicians should remain vigilant and closely monitor the treatment response in this group of patients and promptly take relevant actions (extending the duration of treatment or changing the regimen).
Implementation considerations
The high treatment success rates shown for the BDLLfxC regimen in the BEAT-TB trial and favourable comparison with the current SoC regimens led to an overall recommendation for implementation under routine programmatic conditions. Given that this recommendation is conditional, ongoing operational research will be needed, to add further knowledge that can be used to adjust and improve implementation guidance for the regimen (8).
Patient selection
Eligibility for the BDLLfxC regimen should generally align with the criteria outlined in the BEAT-TB trial. Once patients are started on treatment, providing effective support is crucial to ensure treatment adherence. Close monitoring (for AEs, responses to treatment and developing drug resistance) is essential, as are proper management of side effects and prevention of complications from drug– drug interactions.
Drug susceptibility testing
A comprehensive history of prior exposure to the regimen’s components and the outcomes of previous episodes of TB should be taken. Prior loss to the programme while on a bedaquiline-containing regimen, particularly if the culture is still positive when this occurs, increases the risk of developing bedaquiline resistance (52).
DST for FQ is strongly advised, although it should not delay the initiation of the BDLLfxC. When DST results confirm FQ susceptibility, clofazimine should be stopped, then BDLLfx can continue. When DST results confirm FQ resistance, levofloxacin should be stopped, then BDLC can continue. If DST is not done or is unavailable, then all five drugs should be given.
There is an opportunity to improve global capacity for DST for bedaquiline, delamanid and linezolid, given that current resources are limited and no rapid molecular tests are available for these medications. Prompt treatment initiation is vital, especially for patients with no prior exposure; this is particularly relevant in countries where bedaquiline usage is low, minimizing the risk of resistance transmission. Individuals with a history of receiving bedaquiline, delamanid (or pretomanid) and linezolid for more than a month should have recent DST results confirming susceptibility before being enrolled on the BDLLfxC regimen. In settings where DST for bedaquiline, delamanid and linezolid can be done and resistance to any of these medicines is confirmed, the regimens should not be used.
Drug–drug interactions
It is essential to consider drug–drug interactions when administering TB and HIV medications in combination. The preferred ART regimens for coadministration with BDLLfxC are dolutegravir-based in combination with two nucleoside reverse transcriptase inhibitors (NRTIs). The NRTI zidovudine should be avoided because of the overlapping toxicity of myelosuppression. The non-NRTIs (efavirenz) is contraindicated with bedaquiline. For newer agents, package inserts should be consulted.
Other considerations
There was no exclusion based on BMI. Eighty-five (42%) patients randomized to BDLLfxC had a BMI below 18.5 g/m² . No major effect of a low BMI was noted on the efficacy of the BDLLfxC versus the control (RD –4.1% (95% CI: –16.5%, 8.3%), but the evidence is too uncertain to draw specific conclusions.
In the BEAT-TB trial patients with severe anaemia (haemoglobin <8 g/dL) could receive a transfusion before starting treatment in either group. If the person’s haemoglobin levels recovered to above 8 g/dL, they could then be enrolled in the trial. In programmatic settings, if transfusion is unavailable or considered too risky, the treating physician must consider the benefit–risk profile of starting treatment with the linezolid-containing BDLLfxC regimen versus the 9-month treatment regimen that does not include linezolid or a longer individualized regimen that is linezolid sparing.
Some groups of patients were excluded from the BEAT-TB trial, such as those with liver enzyme measurements three or more times over the upper limit of normal, people with a corrected QT interval by Fredericia (QTcF) of more than 480 ms, or with a current peripheral neuropathy of Grade 3–4. These groups of patients may still receive the regimen if the treating physician judges it to be the best option despite these contraindications and where adequate monitoring is available.
Regimen composition, dosing of component medicines and frequency
The dosing frequency should be 7 days a week. The BDLLfxC regimen comprises bedaquiline, delamanid and linezolid with levofloxacin and/or clofazimine throughout the duration of the regimen, depending on quinolone susceptibility. Delamanid was administered twice daily for 8 weeks and then daily for 16 weeks.
Bedaquiline dosing schemes
The dosing strategy used in the BEAT-TB trial was daily as a loading dose of 400mg for 2 weeks, then 200mg three times weekly for 22 weeks. As an alternative, it is possible to use the daily dosing strategy (i.e. loading dose daily of 200) for the first 2 months and then continue daily 100mg for the next 4 months.
Dosing of linezolid
For adults, the prescribed dose of linezolid was 600 mg for the entire duration of therapy, without any planned dose reductions. These patients continued their treatment with BDLfx or BDLfxC. Clinicians assessed the response to treatment, and if they determined that the response was satisfactory, the patients were allowed to continue with their treatment regimen.
Regimen duration, changes and extensions
Treatment may be extended to nine months if culture conversion is not sustained by week 16. In the BEAT-TB trial, the regimen was prolonged for three patients because of late culture conversion. All possible efforts should be made to support the patient and manage the AEs to ensure uninterrupted treatment and the intake of all medicines in the regimen; however, when medicine cannot be tolerated, it should be stopped.
Linezolid is the leading cause of most AEs. If the clinician determines that the patient has responded well to treatment and that continuing therapy with linezolid would not be in the patient’s best interest, permanent discontinuation of linezolid may occur.
Of the patients studied, 159 (78.7%) completed the entire 6-month course of BDLLfxC without interruptions. Eighteen patients had their linezolid treatment permanently discontinued owing to AEs: three patients experienced anemia, six developed optic neuritis and nine had peripheral neuropathy. The time of linezolid discontinuation was earliest at 6 weeks and latest at 22 weeks. The BDLLfxC regimen has been studied as a standardized course of treatment. Therefore, modifying the regimen through early discontinuation or replacement of any component medicines, except linezolid-related adverse events, is not advised because it may result in different (and possibly worse) treatment outcomes.
Missing doses and tolerances for treatment interruptions
Linezolid could be temporarily or permanently interrupted but the other medicines are to be used throughout the treatment duration. Ideally, missing doses of drugs due to adverse events or nonadherence in the regimen should be avoided. If doses of all medicines are missed, any consecutive interruption of 7 days or more but less than one month should be made up for by extending the treatment duration (for the number of missed doses).
21 See subgroup considerations.
22 Data on the use of pretomanid in pregnant women are limited. Animal studies do not indicate direct or indirect harmful effects with respect to embryo-fetal development.
23 Additional details on linezolid dosing and possible dose reductions are given in the implementation considerations.
24 Available at https://clinicaltrials.gov/ct2/home.
25 Trial protocol available at https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-022-06331-8
26 Several participants excluded in each dataset due to unconfirmed rifampicin resistance.
27 As presented in the expert review (by Dr J-W. Alffenaar, University of Sydney) to the GDG panel in one of the preparatory webinars.
28 Pretomanid has been shown to cause testicular atrophy and impaired fertility in male rats.
29 Estimated regimen prices were calculated using the average weighted price for each medicine (average weighted price accounts for the different prices for each supplier of that medicine weighted by the market share allocation received from each GDF tender), the duration indicated (in months) and assuming 30 days of treatment per month. Actual final costs may differ based on the products delivered.
30 Unpublished, courtesy of Beverley Stringer, Manson unit, Médecins Sans Frontières.
31 In the ZeNix trial, permitted antiretroviral treatments were nevirapine in combination with any nucleoside reverse transcriptase inhibitors (NRTIs); lopinavir/ritonavir in combination with any NRTIs; tenofovir/lamivudine/abacavir (if normal renal function); triple NRTI therapy consisting of zidovudine, lamivudine and abacavir (noting the increased risk of peripheral nerve toxicity with zidovudine and linezolid); and raltegravir in combination with NRTIs.
